

Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions
- PMID: 37277488
- PMCID: PMC10923852
- DOI: 10.1038/s41431-023-01392-y
Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions
Abstract
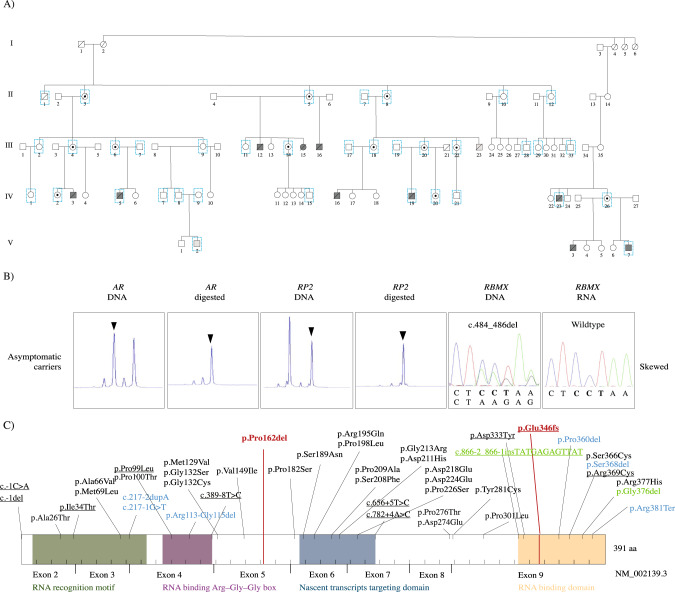
RNA binding motif protein X-linked (RBMX) encodes the heterogeneous nuclear ribonucleoprotein G (hnRNP G) that regulates splicing, sister chromatid cohesion and genome stability. RBMX knock down experiments in various model organisms highlight the gene's importance for brain development. Deletion of the RGG/RG motif in hnRNP G has previously been associated with Shashi syndrome, however involvement of other hnRNP G domains in intellectual disability remain unknown. In the current study, we present the underlying genetic and molecular cause of Gustavson syndrome. Gustavson syndrome was first reported in 1993 in a large Swedish five-generation family presented with profound X-linked intellectual disability and an early death. Extensive genomic analyses of the family revealed hemizygosity for a novel in-frame deletion in RBMX in affected individuals (NM_002139.4; c.484_486del, p.(Pro162del)). Carrier females were asymptomatic and presented with skewed X-chromosome inactivation, indicating silencing of the pathogenic allele. Affected individuals presented minor phenotypic overlap with Shashi syndrome, indicating a different disease-causing mechanism. Investigation of the variant effect in a neuronal cell line (SH-SY5Y) revealed differentially expressed genes enriched for transcription factors involved in RNA polymerase II transcription. Prediction tools and a fluorescence polarization assay imply a novel SH3-binding motif of hnRNP G, and potentially a reduced affinity to SH3 domains caused by the deletion. In conclusion, we present a novel in-frame deletion in RBMX segregating with Gustavson syndrome, leading to disturbed RNA polymerase II transcription, and potentially reduced SH3 binding. The results indicate that disruption of different protein domains affects the severity of RBMX-associated intellectual disabilities.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Comment in
-
Comment on Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions.Eur J Hum Genet. 2024 Mar;32(3):253-256. doi: 10.1038/s41431-023-01498-3. Epub 2023 Nov 29. Eur J Hum Genet. 2024. PMID: 38017187 Free PMC article.
Similar articles
-
Comment on Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions.Eur J Hum Genet. 2024 Mar;32(3):253-256. doi: 10.1038/s41431-023-01498-3. Epub 2023 Nov 29. Eur J Hum Genet. 2024. PMID: 38017187 Free PMC article.
-
Deletion of RBMX RGG/RG motif in Shashi-XLID syndrome leads to aberrant p53 activation and neuronal differentiation defects.Cell Rep. 2021 Jul 13;36(2):109337. doi: 10.1016/j.celrep.2021.109337. Cell Rep. 2021. PMID: 34260915
-
The RBMX gene as a candidate for the Shashi X-linked intellectual disability syndrome.Clin Genet. 2015 Oct;88(4):386-90. doi: 10.1111/cge.12511. Epub 2014 Dec 5. Clin Genet. 2015. PMID: 25256757
-
Novel domains in the hnRNP G/RBMX protein with distinct roles in RNA binding and targeting nascent transcripts.Nucleus. 2010 Jan-Feb;1(1):109-22. doi: 10.4161/nucl.1.1.10857. Nucleus. 2010. PMID: 21327109 Free PMC article.
-
RBMX family proteins connect the fields of nuclear RNA processing, disease and sex chromosome biology.Int J Biochem Cell Biol. 2019 Mar;108:1-6. doi: 10.1016/j.biocel.2018.12.014. Epub 2018 Dec 26. Int J Biochem Cell Biol. 2019. PMID: 30593955 Review.
Cited by
-
Solving medical mysteries with genomics.Eur J Hum Genet. 2024 Mar;32(3):249-250. doi: 10.1038/s41431-024-01568-0. Eur J Hum Genet. 2024. PMID: 38459175 Free PMC article. No abstract available.
-
Loss-of-function variants in RNA binding motif protein X-linked induce neuronal defects contributing to amyotrophic lateral sclerosis pathogenesis.MedComm (2020). 2024 Sep 10;5(9):e712. doi: 10.1002/mco2.712. eCollection 2024 Sep. MedComm (2020). 2024. PMID: 39263607 Free PMC article.
-
Diagnostic yield of 1000 trio analyses with exome and genome sequencing in a clinical setting.Front Genet. 2025 Jun 20;16:1580879. doi: 10.3389/fgene.2025.1580879. eCollection 2025. Front Genet. 2025. PMID: 40620702 Free PMC article.
-
Genetic Profiling of Polymicrogyria in a South Indian Cohort.Ann Indian Acad Neurol. 2025 May 1;28(3):422-425. doi: 10.4103/aian.aian_1096_24. Epub 2025 May 30. Ann Indian Acad Neurol. 2025. PMID: 40445728 Free PMC article.
-
The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies.Genes (Basel). 2025 Feb 1;16(2):176. doi: 10.3390/genes16020176. Genes (Basel). 2025. PMID: 40004505 Free PMC article.
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
