

Genetic studies of paired metabolomes reveal enzymatic and transport processes at the interface of plasma and urine
- PMID: 37277652
- PMCID: PMC10260405
- DOI: 10.1038/s41588-023-01409-8
Genetic studies of paired metabolomes reveal enzymatic and transport processes at the interface of plasma and urine
Abstract
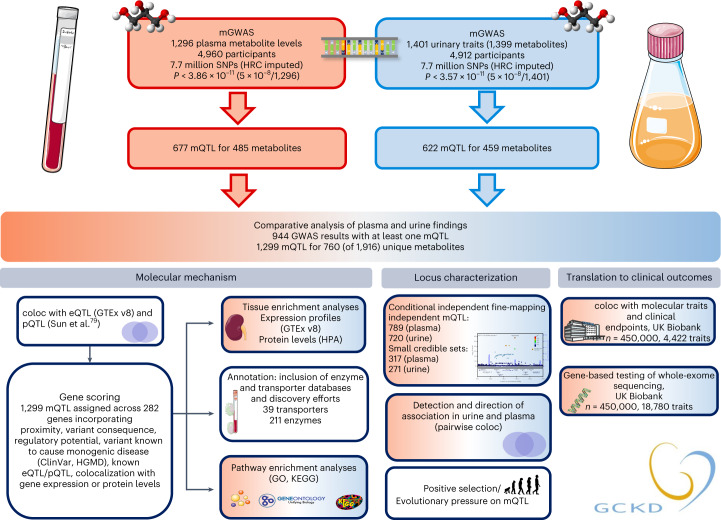
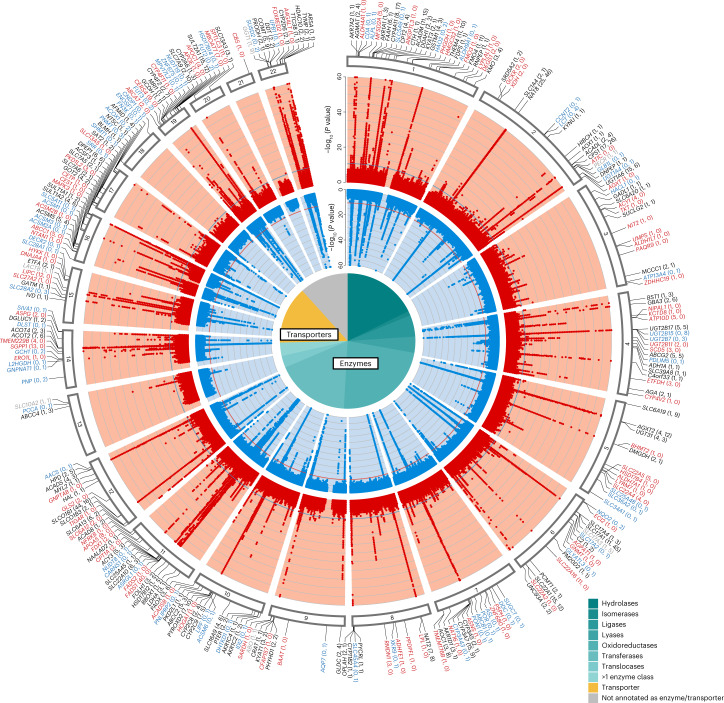
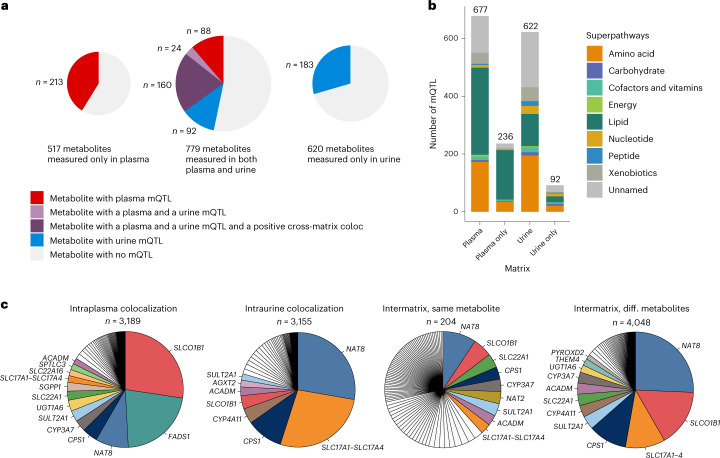
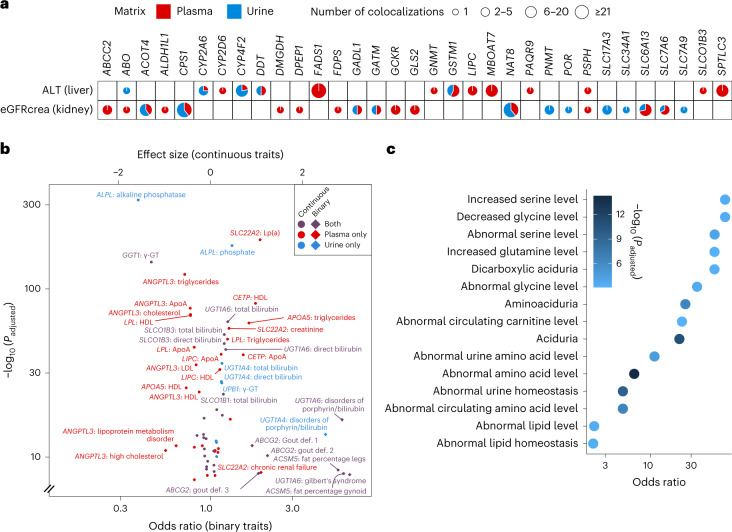
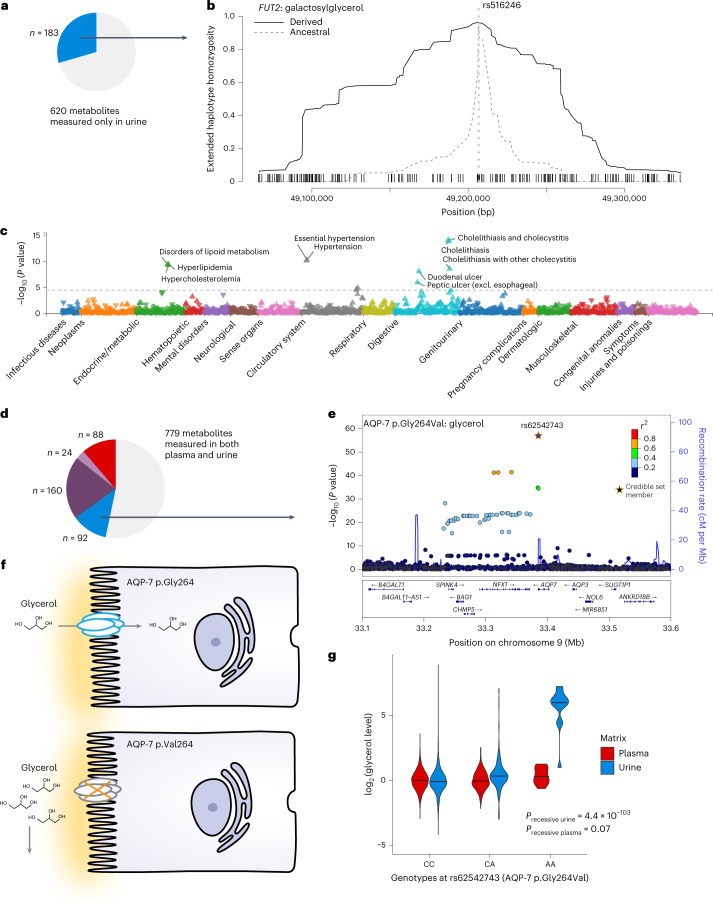
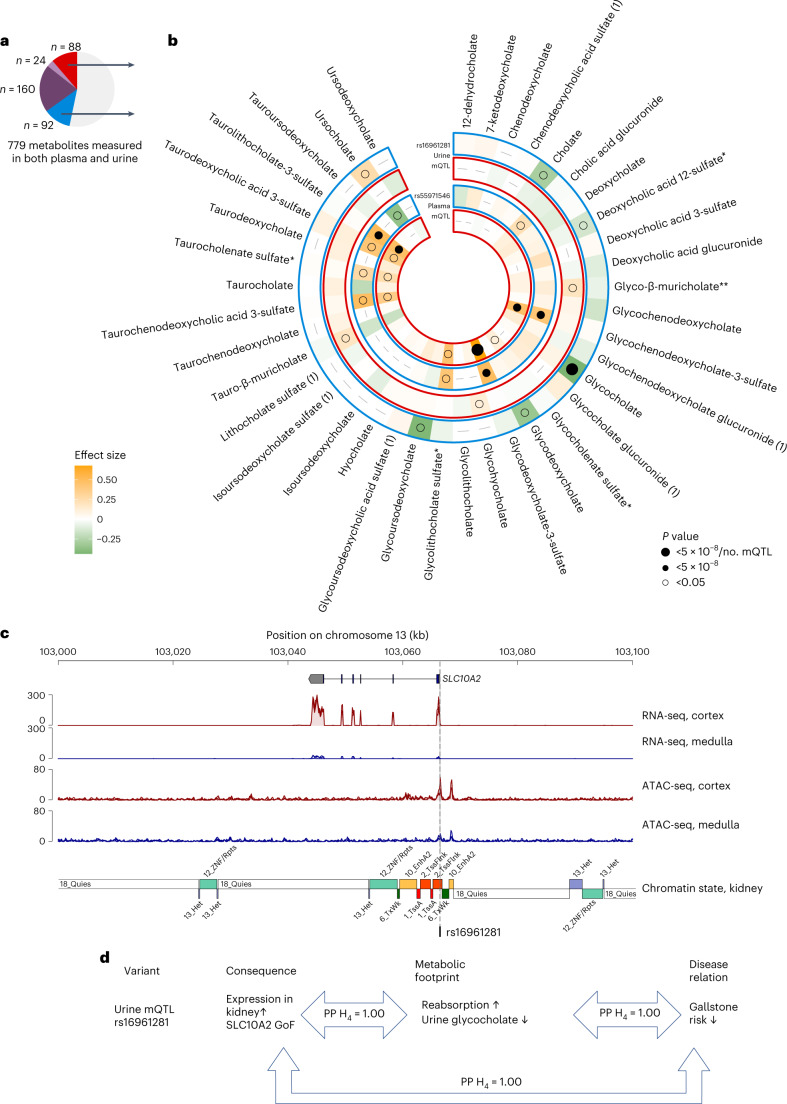
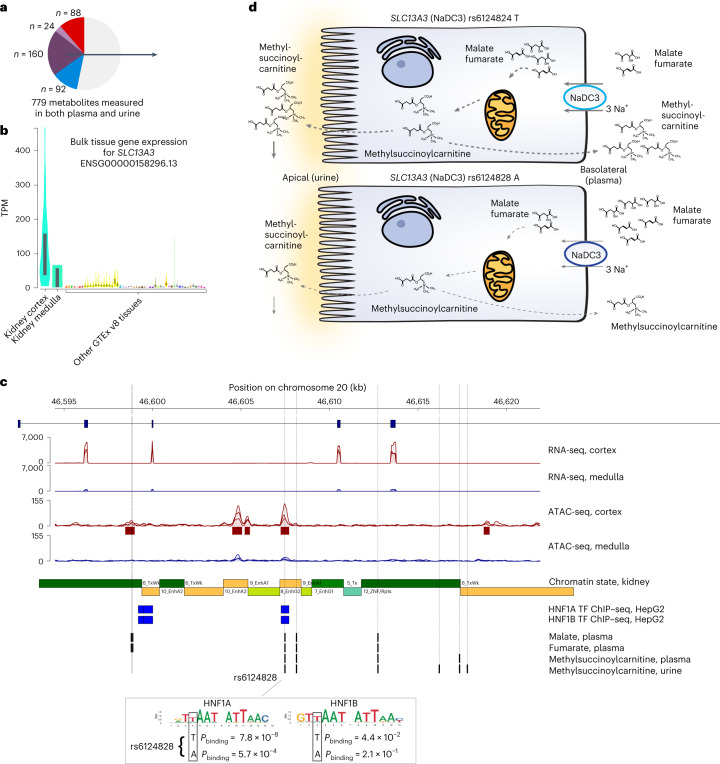
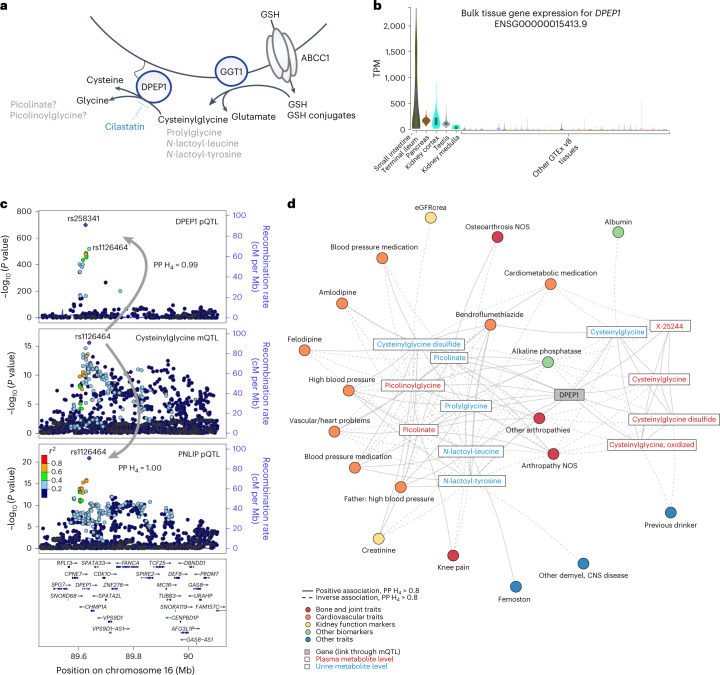
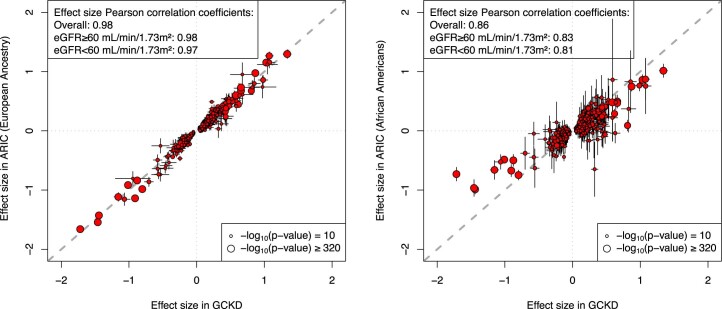
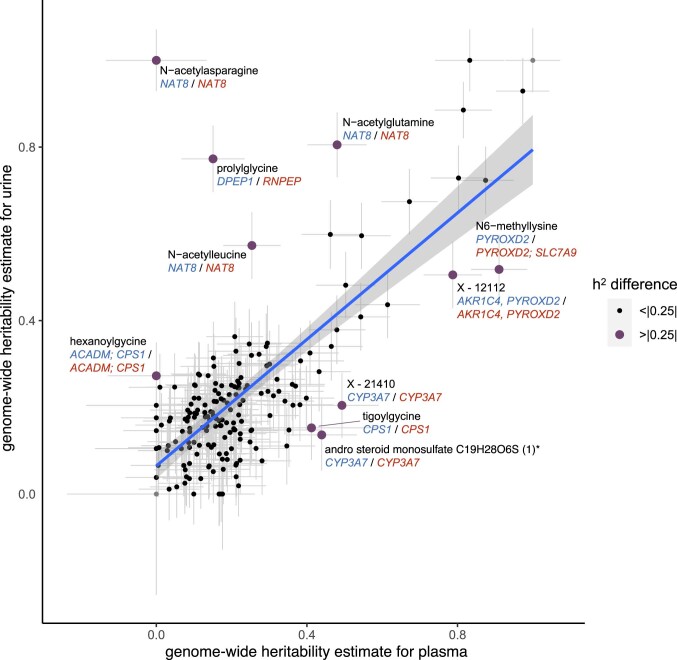
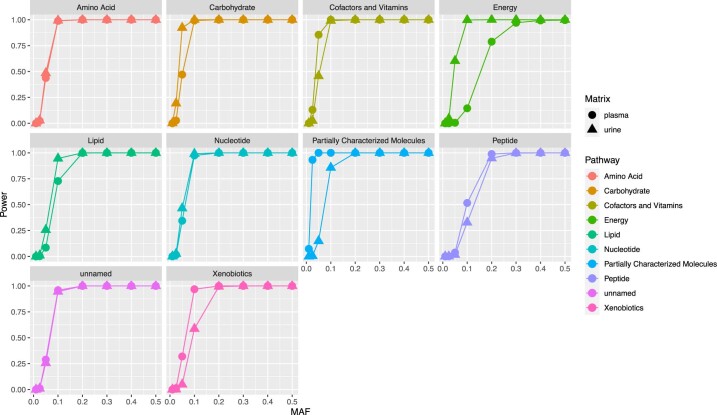
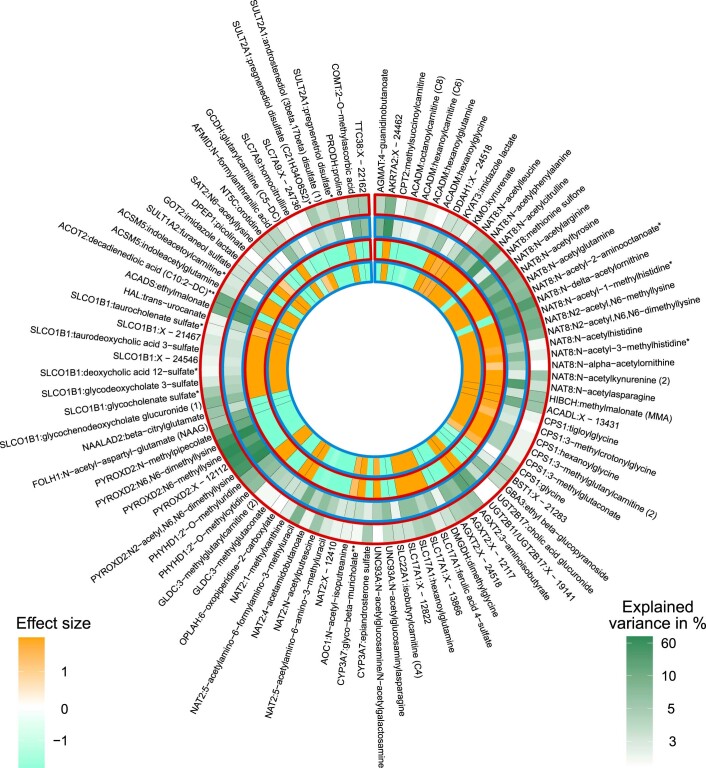
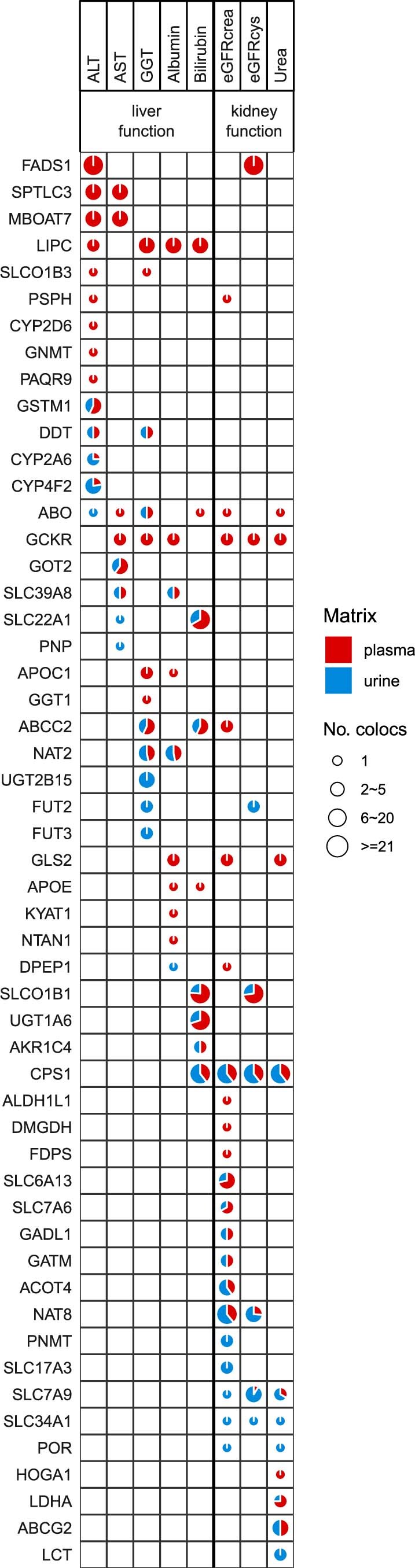
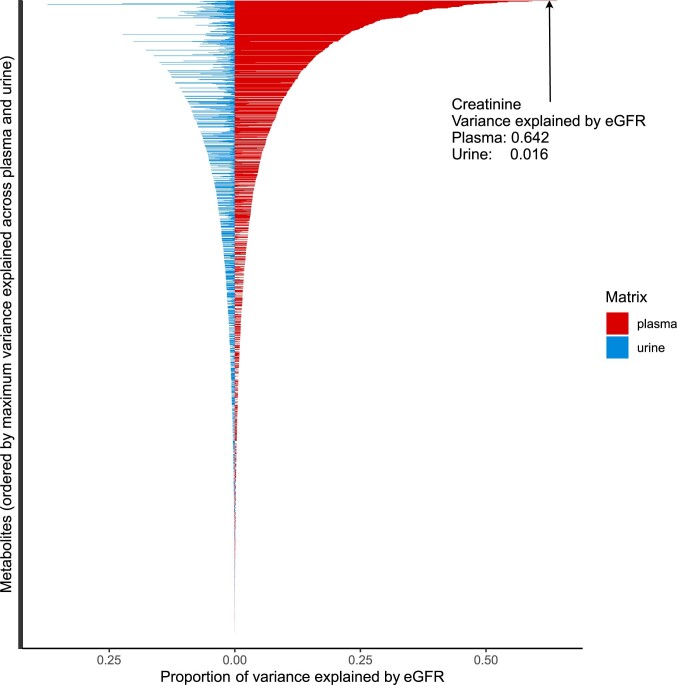
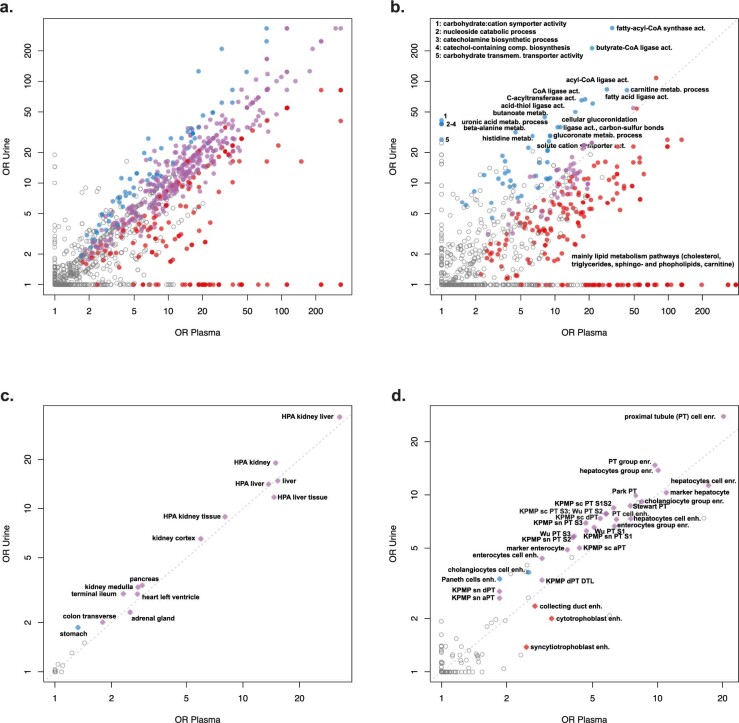
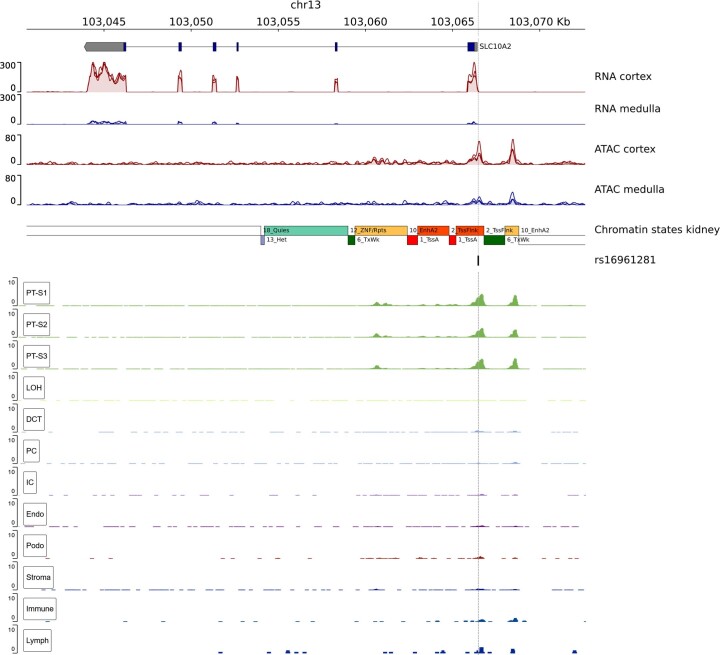
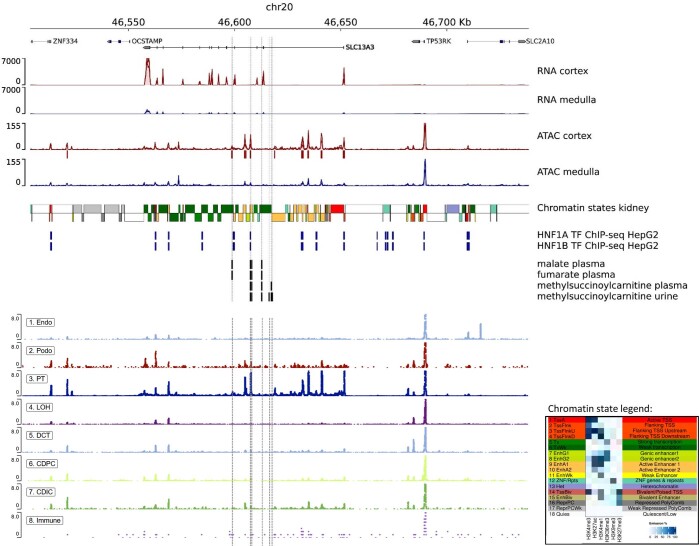
The kidneys operate at the interface of plasma and urine by clearing molecular waste products while retaining valuable solutes. Genetic studies of paired plasma and urine metabolomes may identify underlying processes. We conducted genome-wide studies of 1,916 plasma and urine metabolites and detected 1,299 significant associations. Associations with 40% of implicated metabolites would have been missed by studying plasma alone. We detected urine-specific findings that provide information about metabolite reabsorption in the kidney, such as aquaporin (AQP)-7-mediated glycerol transport, and different metabolomic footprints of kidney-expressed proteins in plasma and urine that are consistent with their localization and function, including the transporters NaDC3 (SLC13A3) and ASBT (SLC10A2). Shared genetic determinants of 7,073 metabolite-disease combinations represent a resource to better understand metabolic diseases and revealed connections of dipeptidase 1 with circulating digestive enzymes and with hypertension. Extending genetic studies of the metabolome beyond plasma yields unique insights into processes at the interface of body compartments.
© 2023. The Author(s).
Conflict of interest statement
R.P.M. and E.D.K. are employees of Metabolon and, as such, have affiliations or financial involvement with Metabolon. J.M. is an employee of Bayer. All other authors declare no competing interests.
Figures
References
-
- Boron, W. F. & Boulpaep, E. L. Medical Physiology (Elsevier, 2017).
-
- Anzai N, Endou H. Urate transporters: an evolving field. Semin. Nephrol. 2011;31:400–409. - PubMed
-
- Evans AM, et al. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics. 2014;4:132.
