

Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis
- PMID: 37277655
- PMCID: PMC10307631
- DOI: 10.1038/s41590-023-01527-9
Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis
Abstract
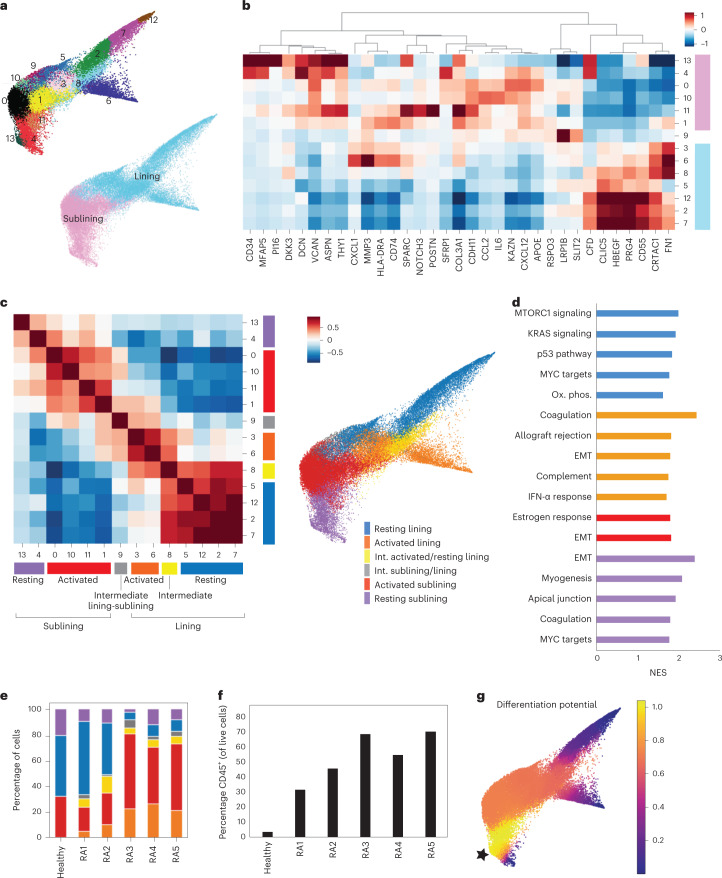
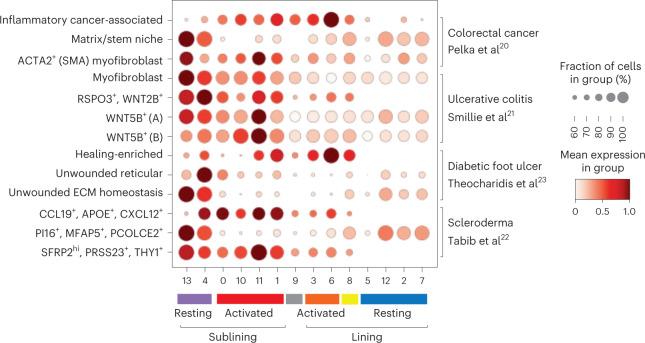
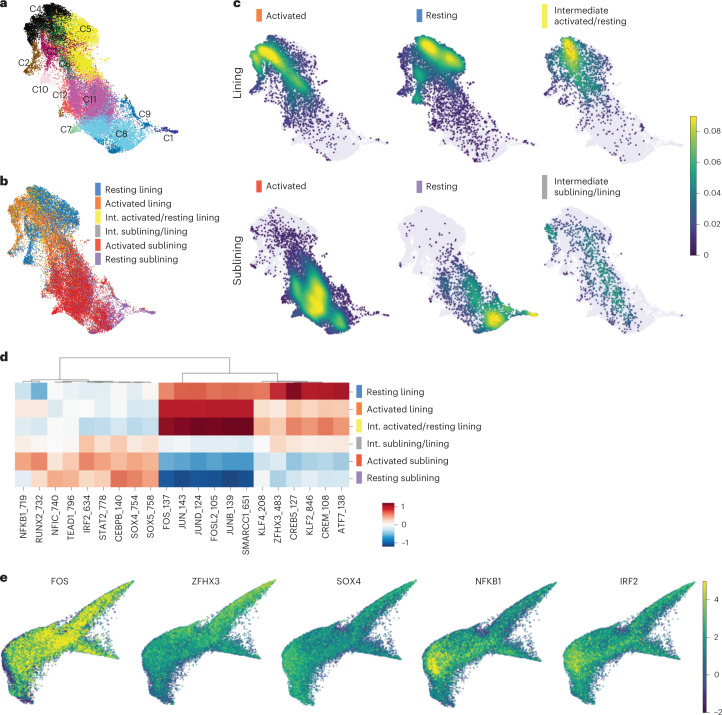
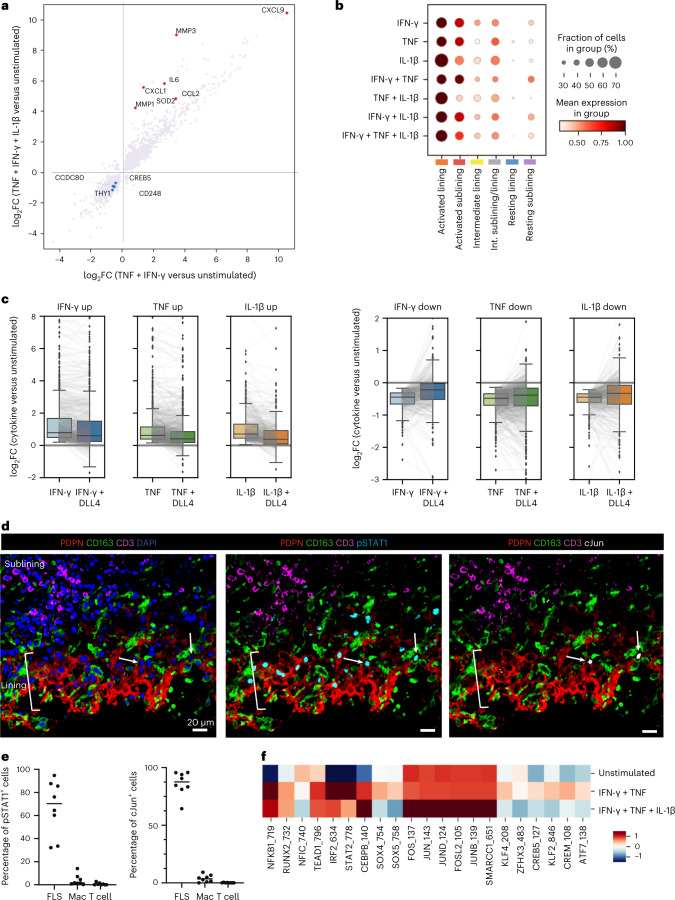
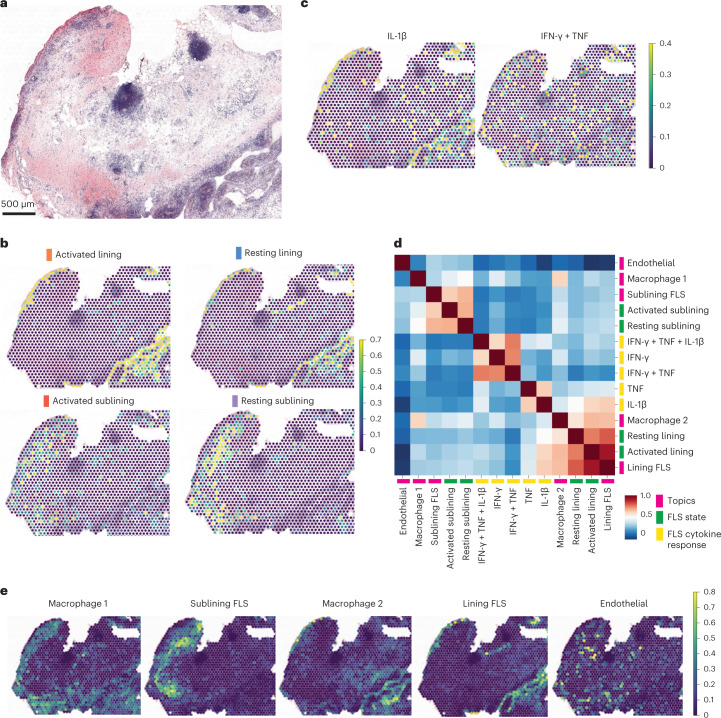
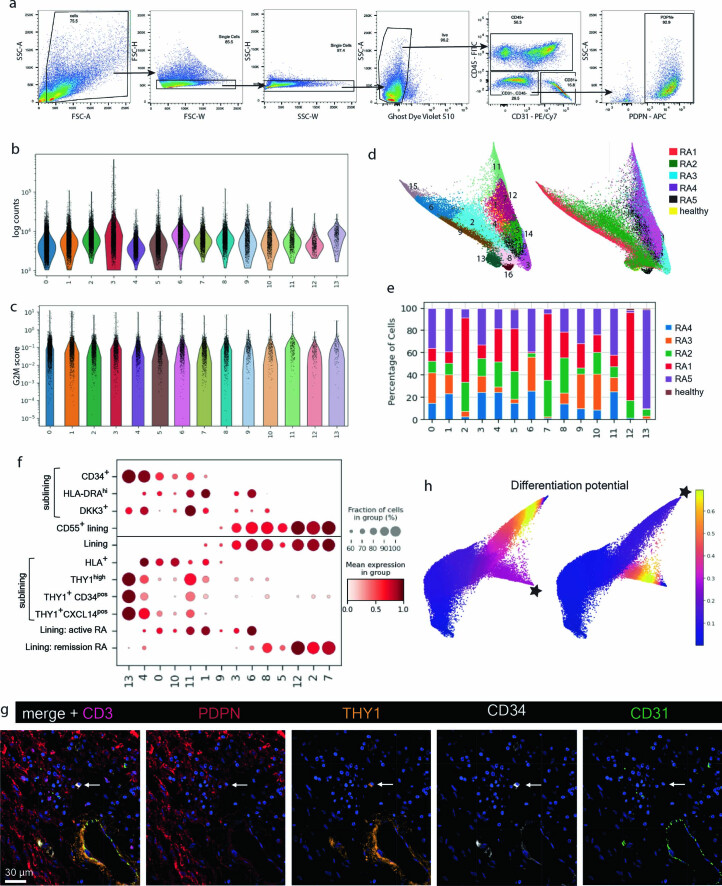
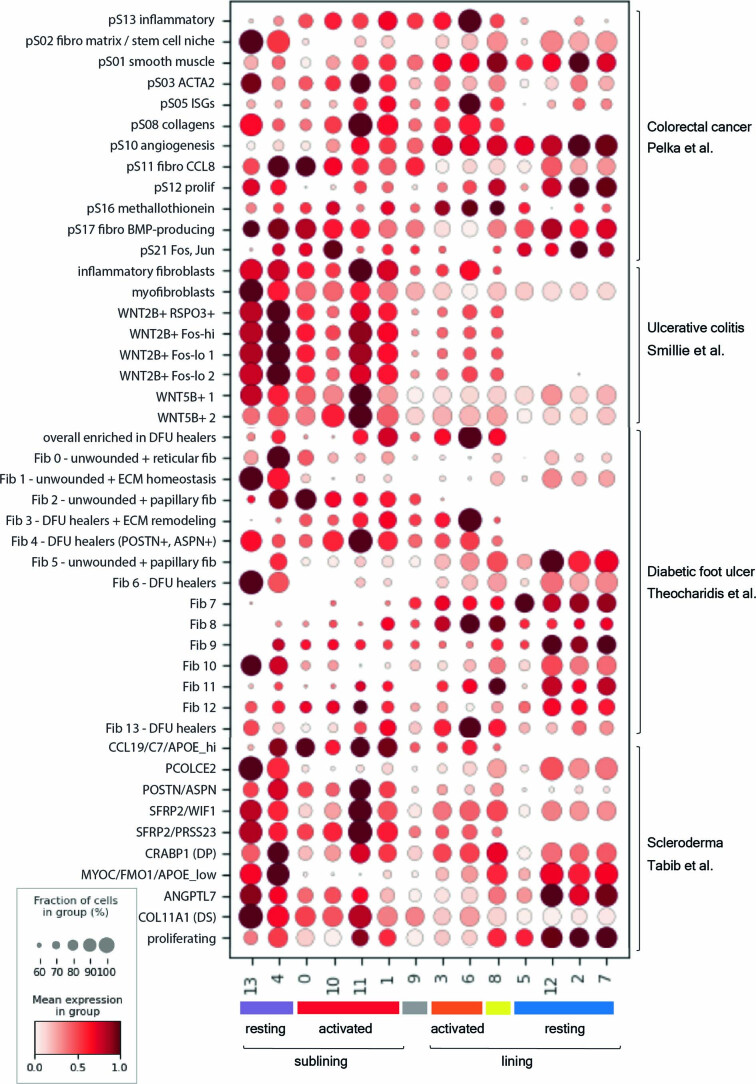
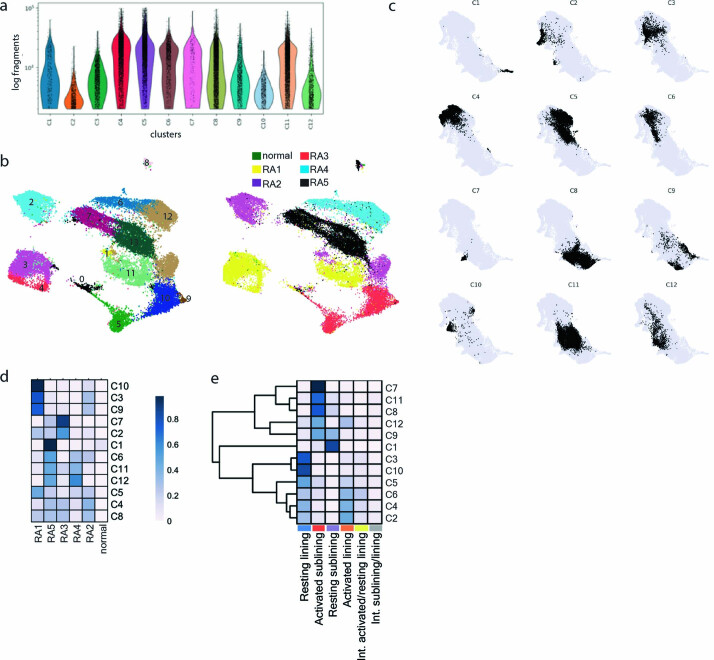
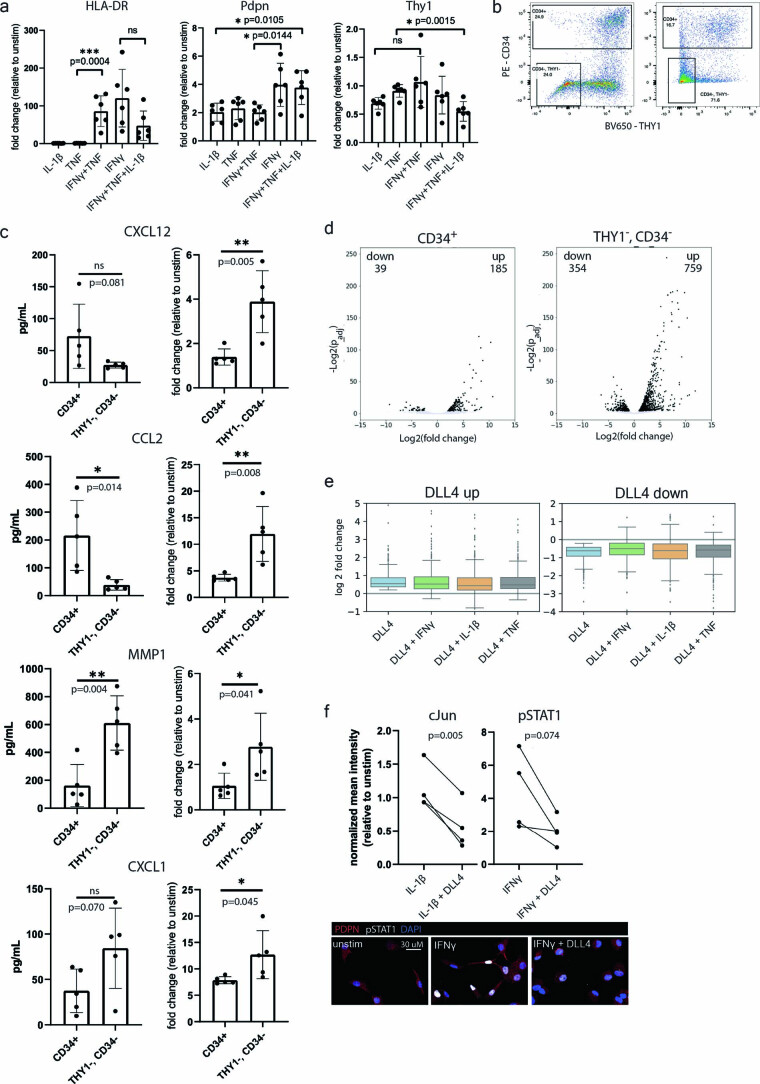
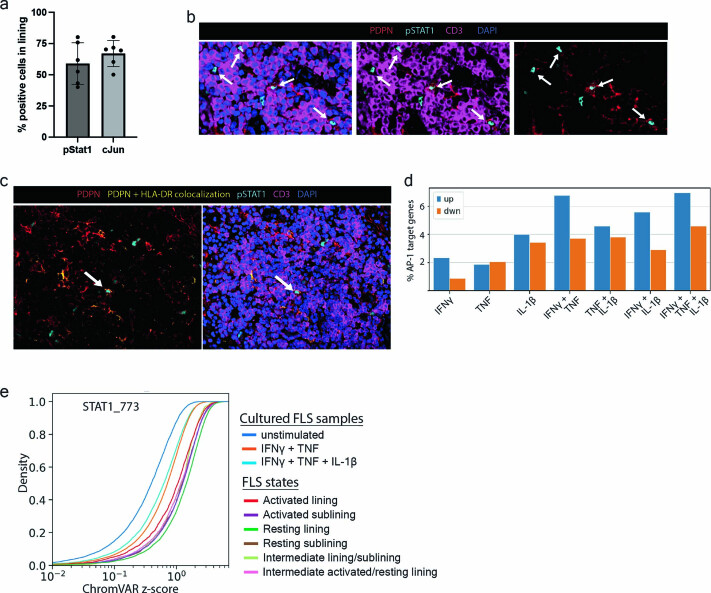
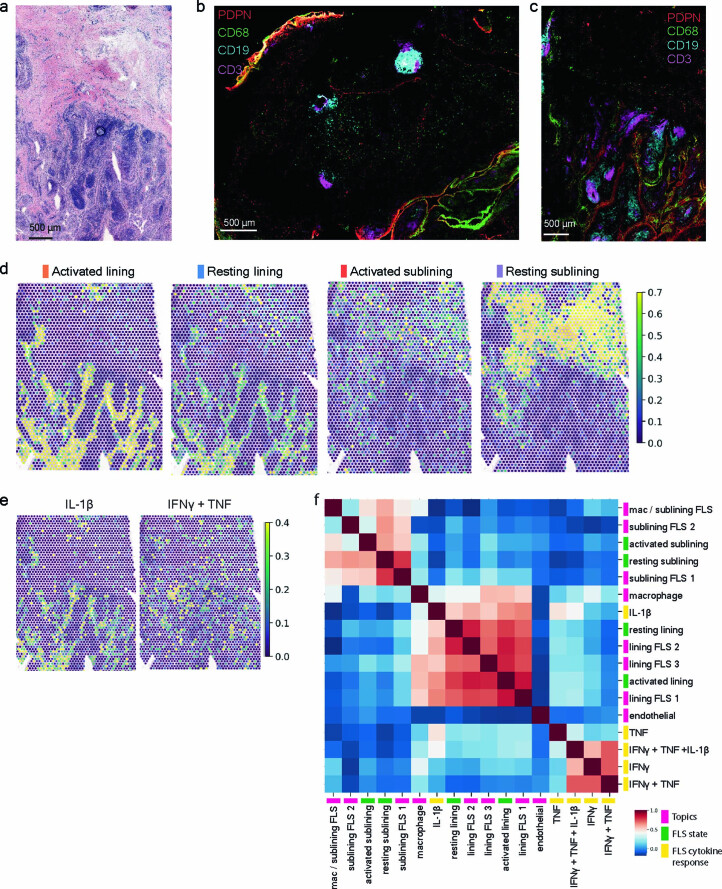
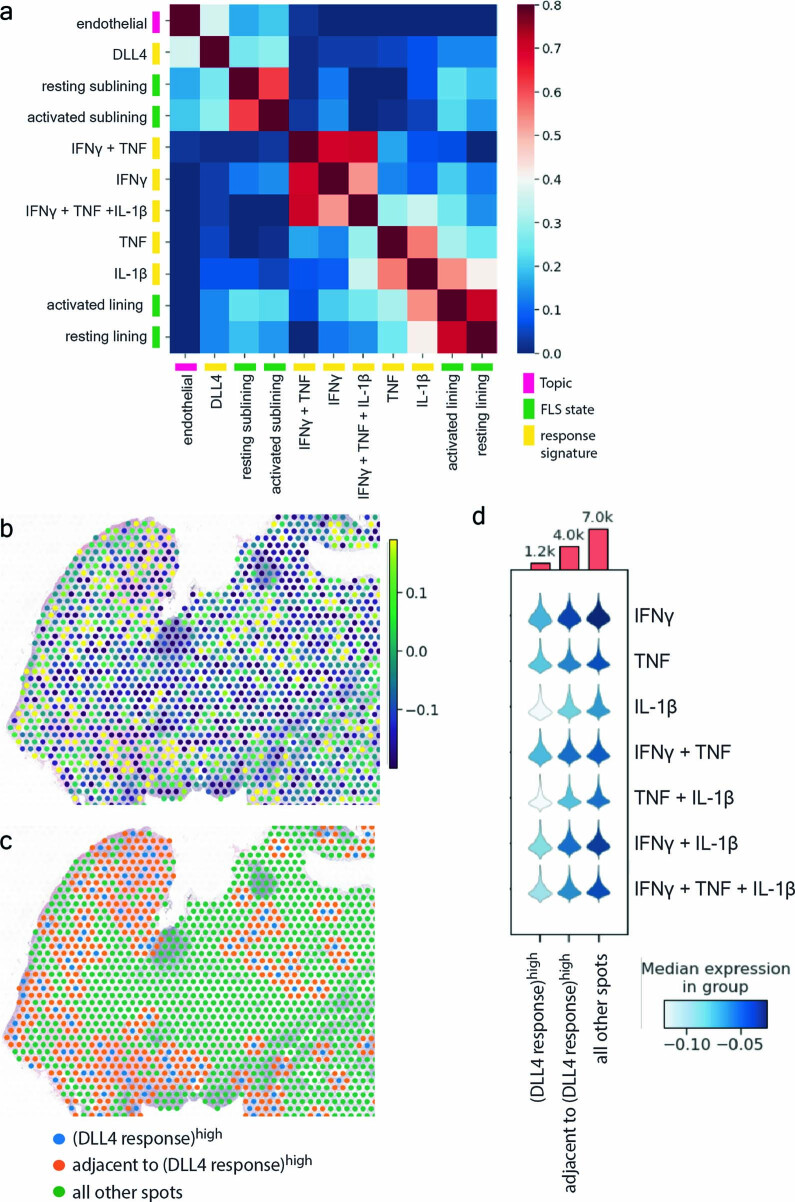
Inflammation of non-barrier immunologically quiescent tissues is associated with a massive influx of blood-borne innate and adaptive immune cells. Cues from the latter are likely to alter and expand activated states of the resident cells. However, local communications between immigrant and resident cell types in human inflammatory disease remain poorly understood. Here, we explored drivers of fibroblast-like synoviocyte (FLS) heterogeneity in inflamed joints of patients with rheumatoid arthritis using paired single-cell RNA and ATAC sequencing, multiplexed imaging and spatial transcriptomics along with in vitro modeling of cell-extrinsic factor signaling. These analyses suggest that local exposures to myeloid and T cell-derived cytokines, TNF, IFN-γ, IL-1β or lack thereof, drive four distinct FLS states some of which closely resemble fibroblast states in other disease-affected tissues including skin and colon. Our results highlight a role for concurrent, spatially distributed cytokine signaling within the inflamed synovium.
© 2023. The Author(s).
Conflict of interest statement
A.Y.R. is an SAB member and has equity in Sonoma Biotherapeutics, Santa Ana Bio, RAPT Therapeutics and Vedanta Biosciences. He is an SEB member of Amgen and BioInvent and is a co-inventor or has IP licensed to Takeda that is unrelated to the content of the present study. S.M.G. consults for UCB and has research support from Novartis both of which are unrelated to the present study. The remaining authors declare no competing interests.
Figures
Comment in
-
Fibroblast multiplicity in RA: a synovial state of affairs.Nat Rev Rheumatol. 2023 Oct;19(10):609-610. doi: 10.1038/s41584-023-01015-2. Nat Rev Rheumatol. 2023. PMID: 37605002 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
