

SUsPECT: a pipeline for variant effect prediction based on custom long-read transcriptomes for improved clinical variant annotation
- PMID: 37280537
- PMCID: PMC10245480
- DOI: 10.1186/s12864-023-09391-5
SUsPECT: a pipeline for variant effect prediction based on custom long-read transcriptomes for improved clinical variant annotation
Abstract
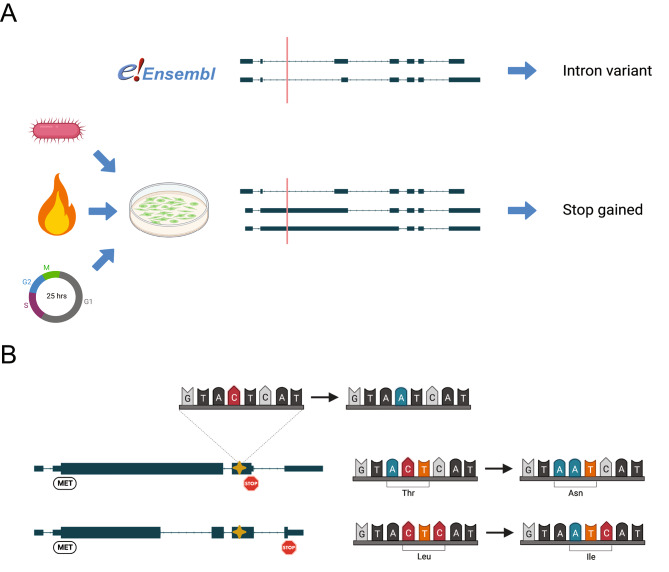
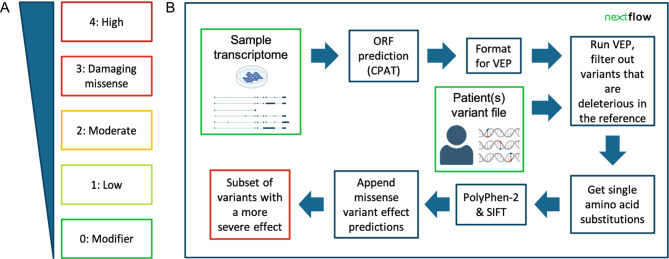
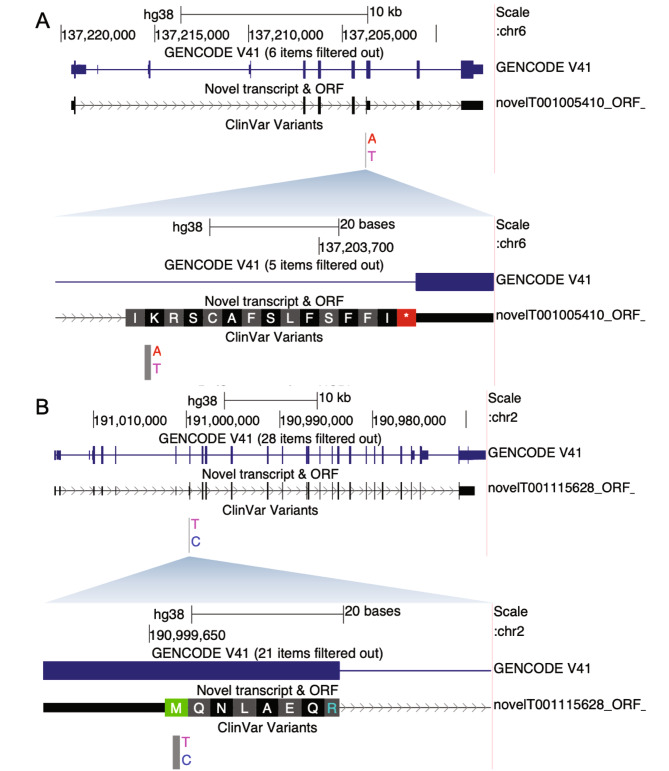
Our incomplete knowledge of the human transcriptome impairs the detection of disease-causing variants, in particular if they affect transcripts only expressed under certain conditions. These transcripts are often lacking from reference transcript sets, such as Ensembl/GENCODE and RefSeq, and could be relevant for establishing genetic diagnoses. We present SUsPECT (Solving Unsolved Patient Exomes/gEnomes using Custom Transcriptomes), a pipeline based on the Ensembl Variant Effect Predictor (VEP) to predict variant impact on custom transcript sets, such as those generated by long-read RNA-sequencing, for downstream prioritization. Our pipeline predicts the functional consequence and likely deleteriousness scores for missense variants in the context of novel open reading frames predicted from any transcriptome. We demonstrate the utility of SUsPECT by uncovering potential mutational mechanisms of pathogenic variants in ClinVar that are not predicted to be pathogenic using the reference transcript annotation. In further support of SUsPECT's utility, we identified an enrichment of immune-related variants predicted to have a more severe molecular consequence when annotating with a newly generated transcriptome from stimulated immune cells instead of the reference transcriptome. Our pipeline outputs crucial information for further prioritization of potentially disease-causing variants for any disease and will become increasingly useful as more long-read RNA sequencing datasets become available.
Keywords: Computational pipeline; Immune response; Medical diagnostics; Primary immunodeficiencies; Rare diseases; Variant effect prediction.
© 2023. The Author(s).
Conflict of interest statement
Not applicable.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
