

Multiple system atrophy - a clinicopathological update
- PMID: 37283673
- PMCID: PMC10209915
- DOI: 10.17879/freeneuropathology-2020-2813
Multiple system atrophy - a clinicopathological update
Abstract
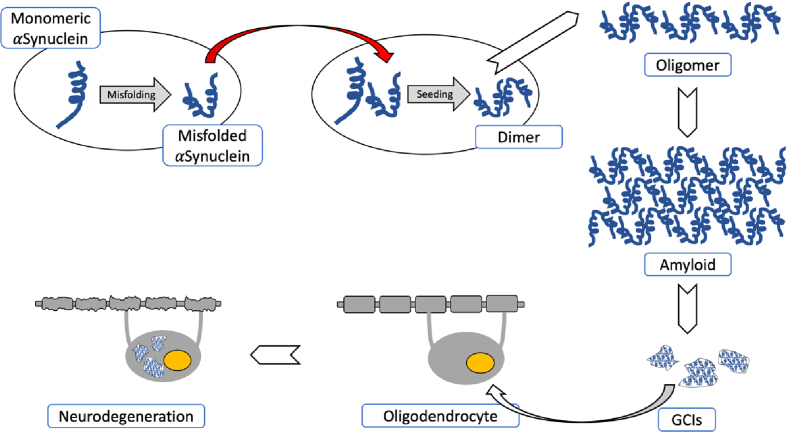
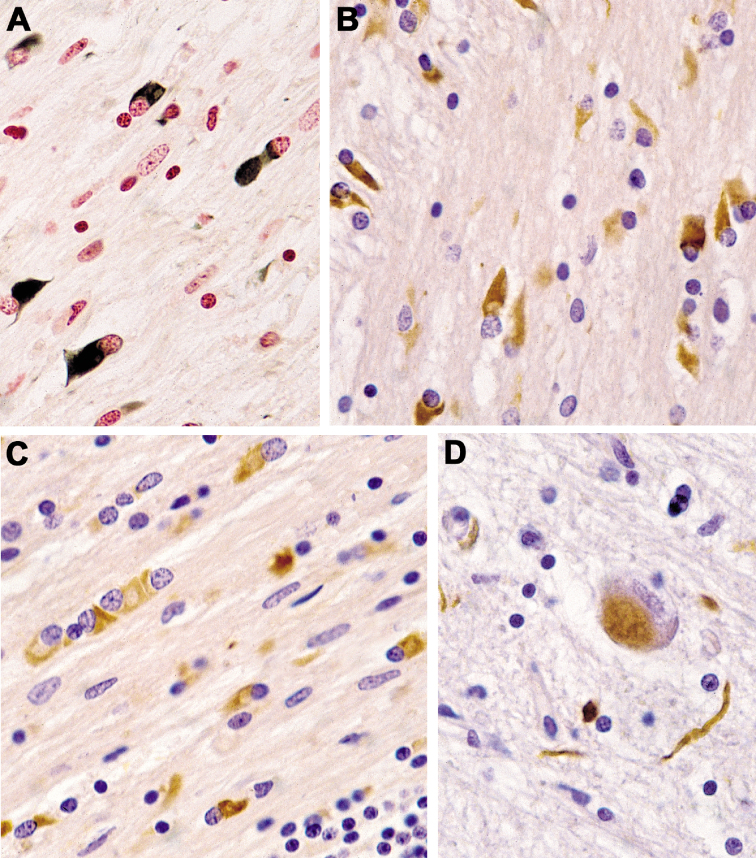
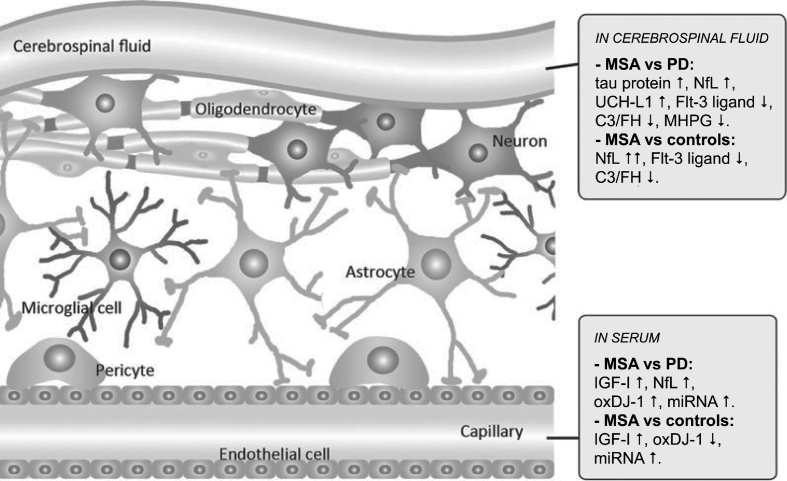
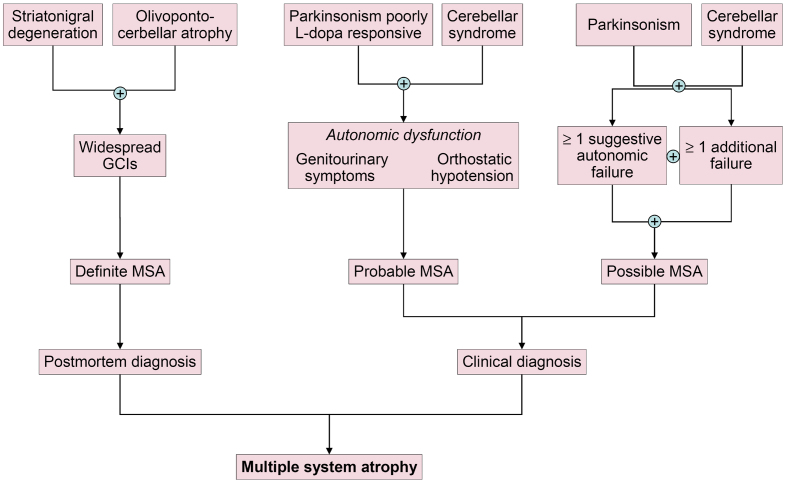
Multiple system atrophy (MSA) is a fatal, adult-onset neurodegenerative disorder of uncertain etiology, clinically characterized by various combinations of Levo-dopa-unresponsive parkinsonism, and cerebellar, motor, and autonomic dysfunctions. MSA is an α-synucleinopathy with specific glioneuronal degeneration involving striatonigral, olivopontocerebellar, autonomic and peripheral nervous systems. The pathologic hallmark of this unique proteinopathy is the deposition of aberrant α-synuclein (αSyn) in both glia (mainly oligodendroglia) and neurons forming pathological inclusions that cause cell dysfunction and demise. The major variants are striatonigral degeneration (MSA with predominant parkinsonism / MSA-P) and olivopontocerebellar atrophy (MSA with prominent cerebellar ataxia / MSA-C). However, the clinical and pathological features of MSA are broader than previously considered. Studies in various mouse models and human patients have helped to better understand the molecular mechanisms that underlie the progression of the disease. The pathogenesis of MSA is characterized by propagation of disease-specific strains of αSyn from neurons to oligodendroglia and cell-to-cell spreading in a "prion-like" manner, oxidative stress, proteasomal and mitochondrial dysfunctions, myelin dysregulation, neuroinflammation, decreased neurotrophic factors, and energy failure. The combination of these mechanisms results in neurodegeneration with widespread demyelination and a multisystem involvement that is specific for MSA. Clinical diagnostic accuracy and differential diagnosis of MSA have improved by using combined biomarkers. Cognitive impairment, which has been a non-supporting feature of MSA, is not uncommon, while severe dementia is rare. Despite several pharmacological approaches in MSA models, no effective disease-modifying therapeutic strategies are currently available, although many clinical trials targeting disease modification, including immunotherapy and combined approaches, are under way. Multidisciplinary research to elucidate the genetic and molecular background of the noxious processes as the basis for development of an effective treatment of the hitherto incurable disorder are urgently needed.
Keywords: Animal models; Biomarkers; Etiopathogenesis; Experimental therapeutics; Glio-neuronal degeneration; Multiple system atrophy; Prion-like seeding; α-synuclein.
Figures
References
-
- Trojanowski JQ, Revesz T (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy, Neuropathol Appl Neurobiol 33:615-20. - PubMed
-
- Jellinger KA, Wenning GK (2016) Multiple system atrophy: pathogenic mechanisms and biomarkers, J Neural Transm (Vienna) 123:555-72. - PubMed
LinkOut - more resources
Full Text Sources