

The Site and Type of CLCN5 Genetic Variation Impact the Resulting Dent Disease-1 Phenotype
- PMID: 37284679
- PMCID: PMC10239918
- DOI: 10.1016/j.ekir.2023.03.012
The Site and Type of CLCN5 Genetic Variation Impact the Resulting Dent Disease-1 Phenotype
Abstract
Introduction: Dent disease is an X-linked recessive disorder associated with low molecular weight proteinuria (LMWP), nephrocalcinosis, kidney stones, and kidney failure in the third to fifth decade of life. It consists of Dent disease 1 (DD1) (60% of patients) because of pathogenic variants in the CLCN5 gene and Dent disease 2 (DD2) with changes in OCRL.
Methods: Retrospective review of 162 patients from 121 different families with genetically confirmed DD1 (82 different pathogenic variants validated using American College of Medical Genetics [ACMG] guidelines). Clinical and genetic factors were compared using observational statistics.
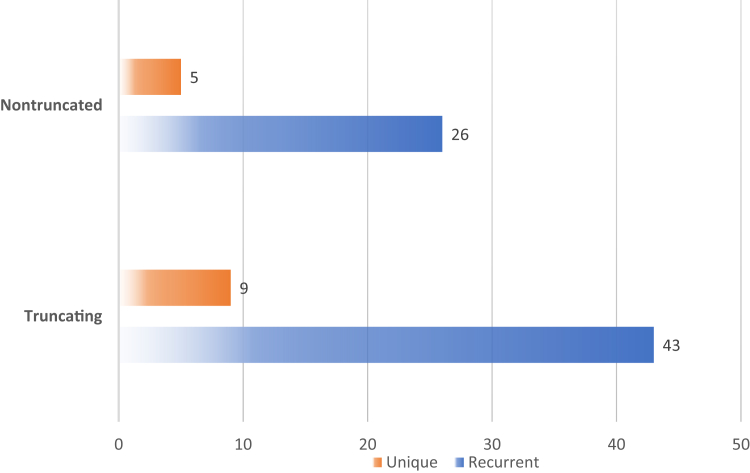
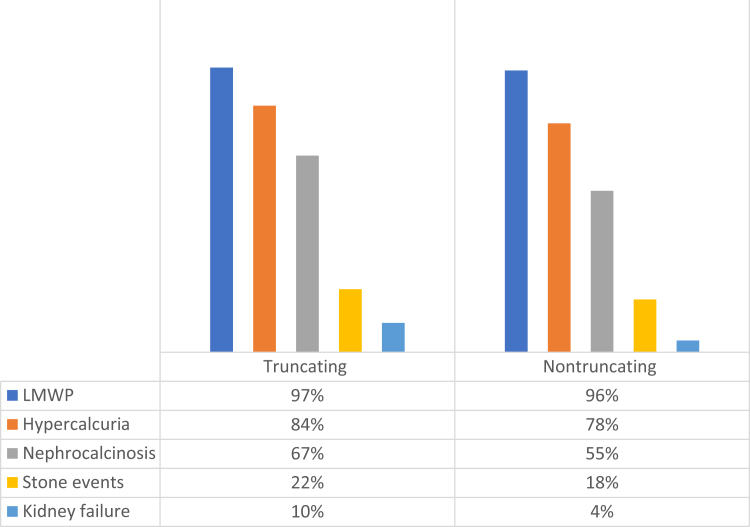
Results: A total of 110 patients had 51 different truncating (nonsense, frameshifting, large deletions, and canonical splicing) variants, whereas 52 patients had 31 different nontruncating (missense, in-frame, noncanonical splicing, and stop-loss) changes. Sixteen newly described pathogenic variants were found in our cohort. Among patients with truncating variants, lifetime stone events positively correlated with chronic kidney disease (CKD) evolution. Patients with truncating changes also experienced stone events earlier in life and manifested a higher albumin excretion rate than the nontruncating group. Nevertheless, neither age of nephrocalcinosis nor CKD progression varied between the truncating versus nontruncating patients. A large majority of nontruncating changes (26/31; 84%) were clustered in the middle exons that encode the voltage ClC domain whereas truncating changes were spread across the protein. Variants associated with kidney failure were restricted to truncating (11/13 cases), plus a single missense variant previously shown to markedly reduce ClC-5 functional activity that was found in the other 2 individuals.
Conclusion: DD1 manifestations, including the risk of kidney stones and progression to kidney failure, may relate to the degree of residual ClC-5 function.
Keywords: CKD; Dent disease; hypercalciuria; nephrocalcinosis; proteinuria.
© 2023 International Society of Nephrology. Published by Elsevier Inc.
Figures





References
-
- Wrong O.M., Norden A.G., Feest T.G. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87:473–493. - PubMed
-
- Lieske J.C., Milliner D.S., Beara-Lasic L., Harris P., Cogal A., Abrash E. In: GeneReviews. Adam M.P., Mirzaa G.M., Pagon R.A., et al., editors. University of Washington; 2012; updated 2017; 1993. Dent disease.
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
