

Epigenetic dysregulation from chromosomal transit in micronuclei
- PMID: 37286593
- PMCID: PMC10322720
- DOI: 10.1038/s41586-023-06084-7
Epigenetic dysregulation from chromosomal transit in micronuclei
Abstract
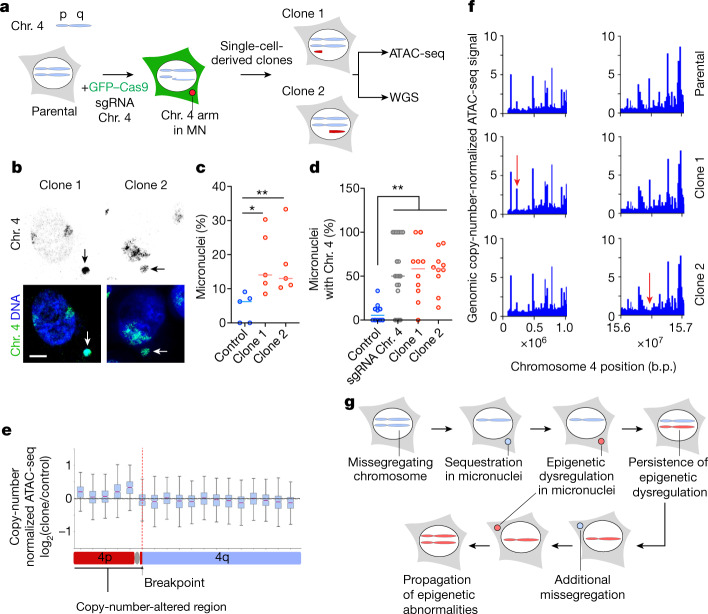
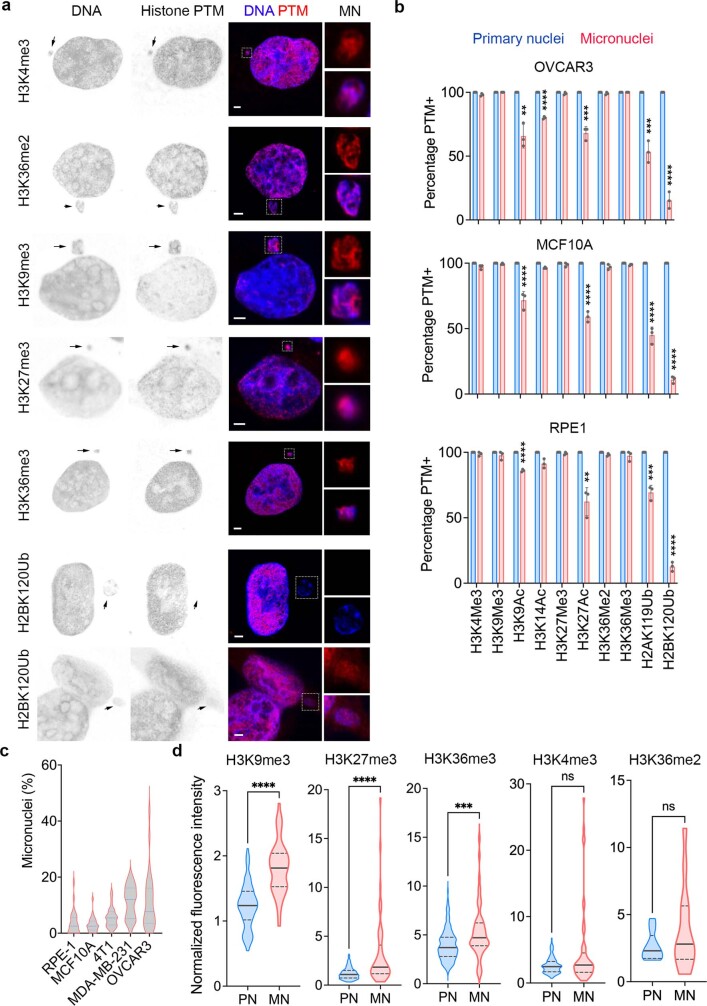
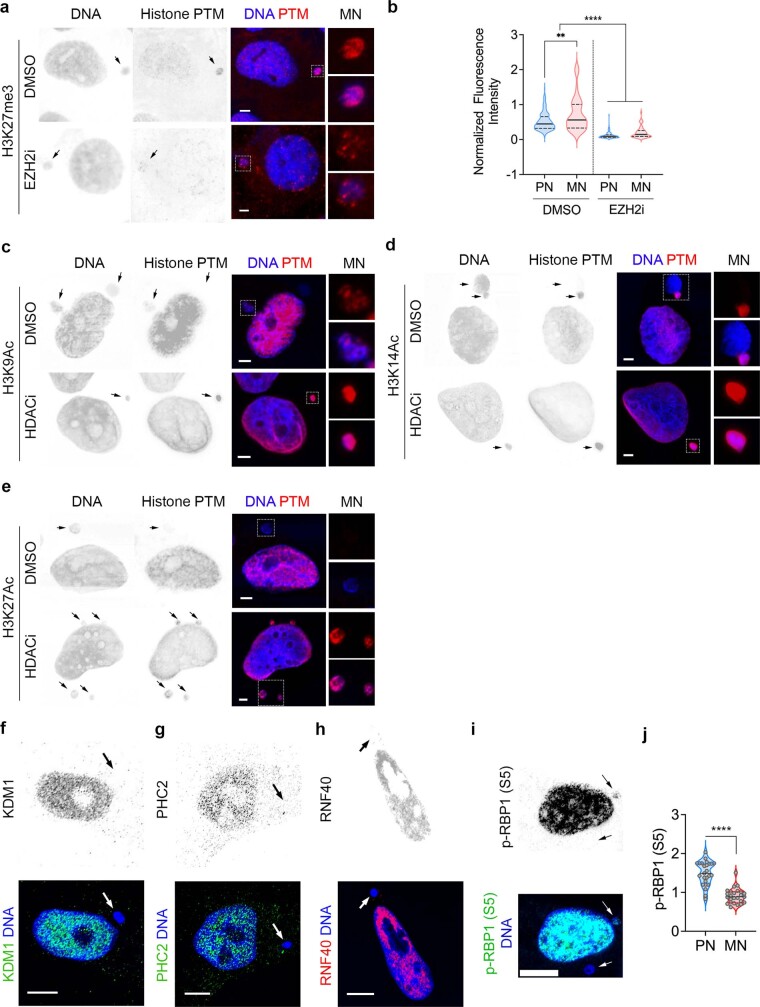
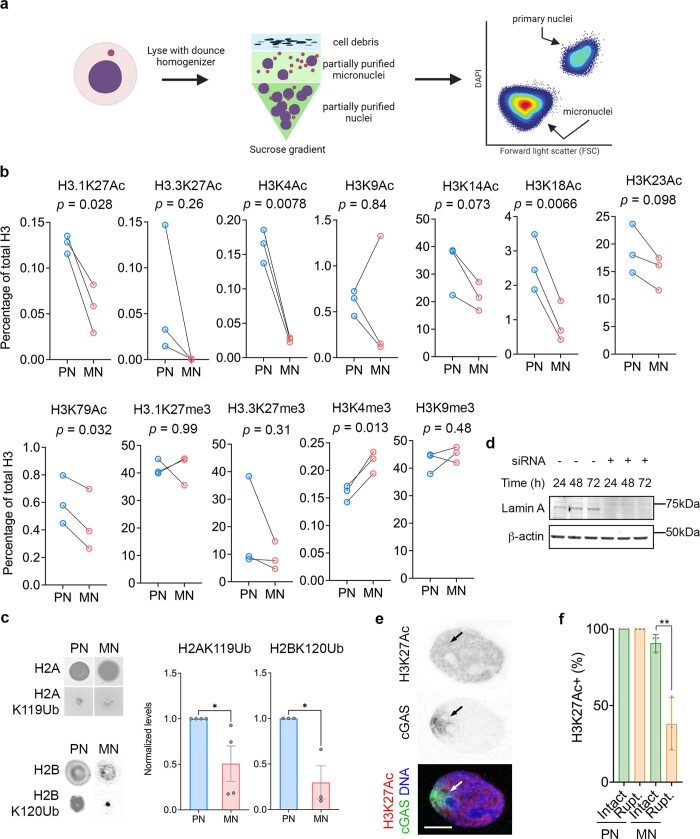
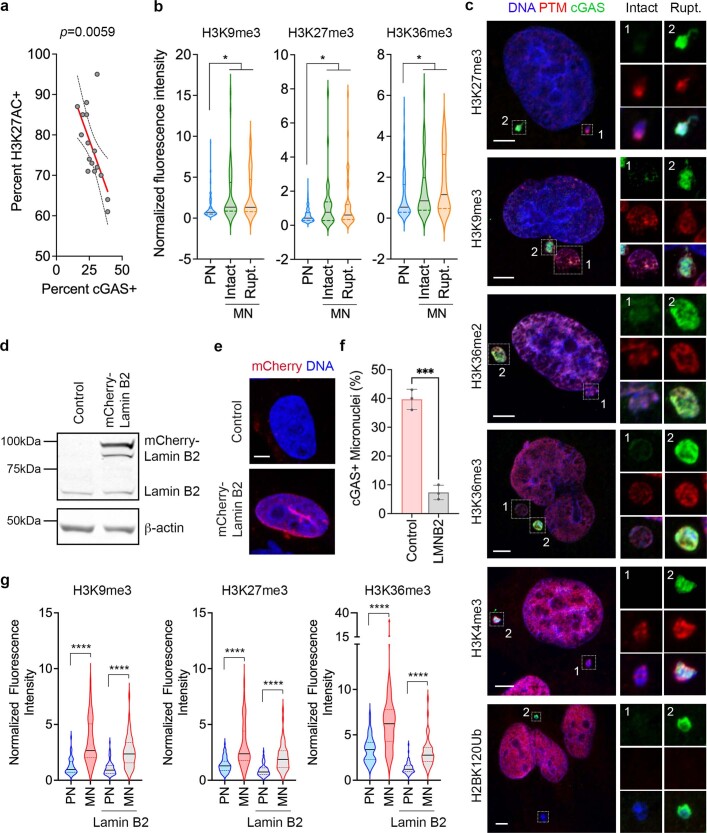
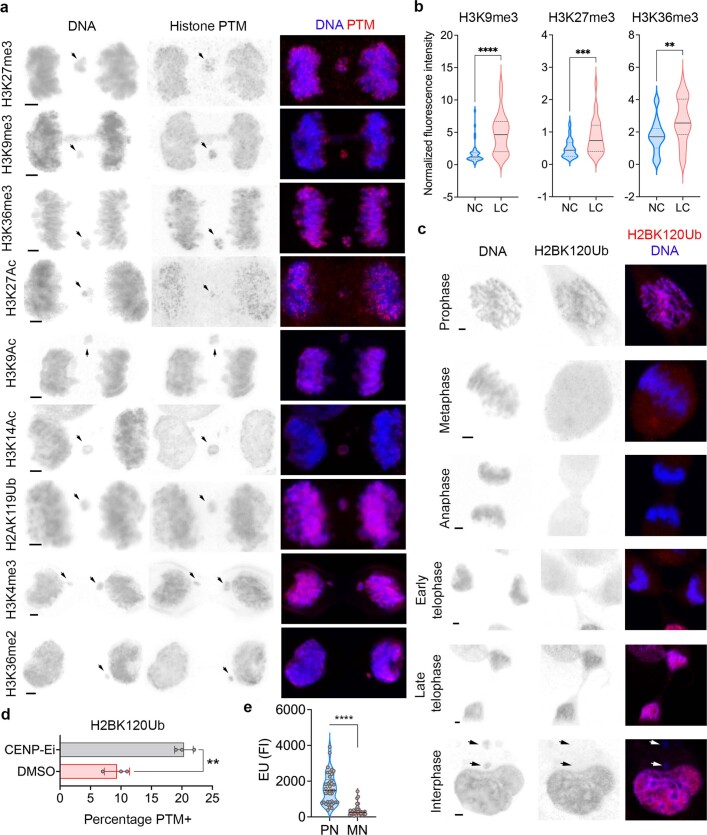
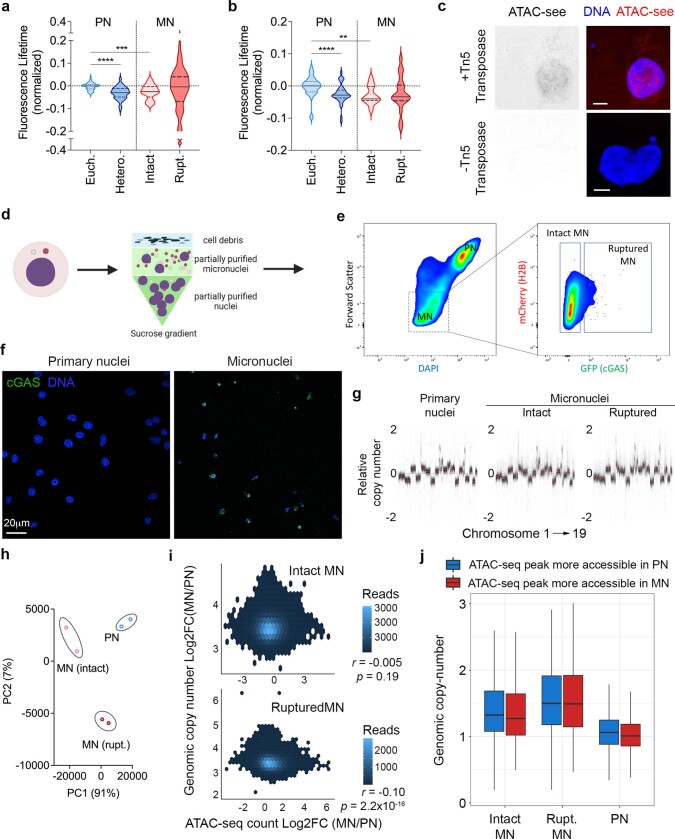
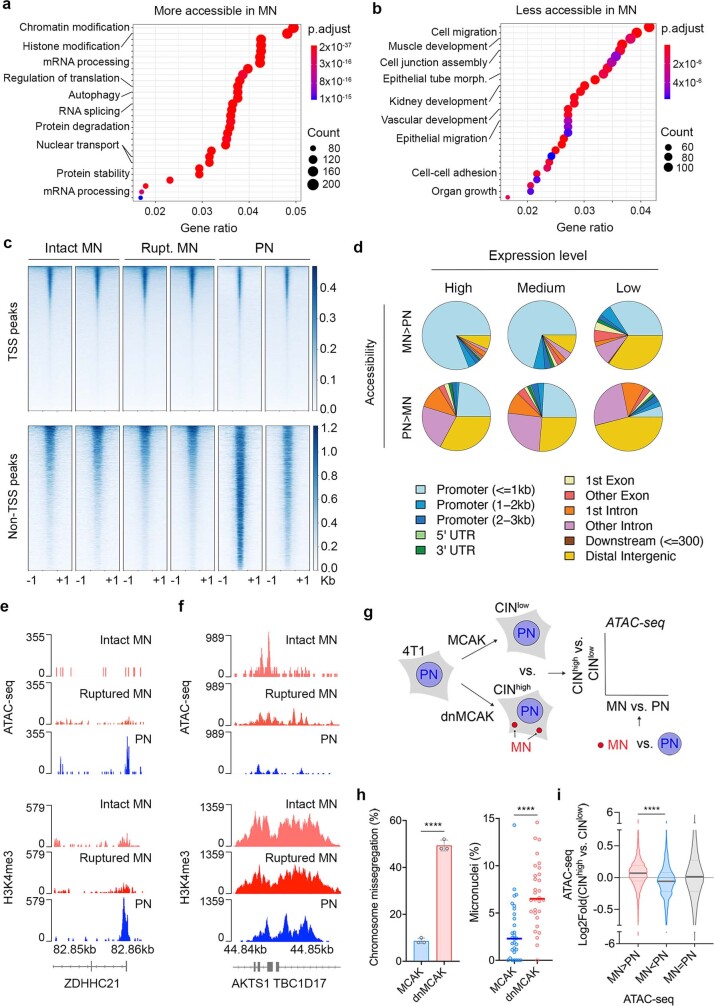
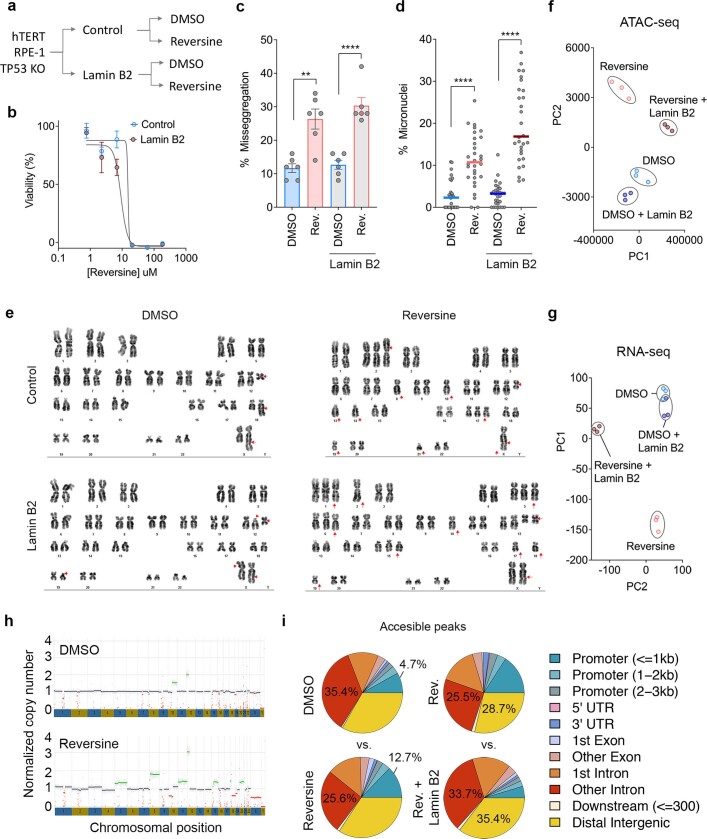
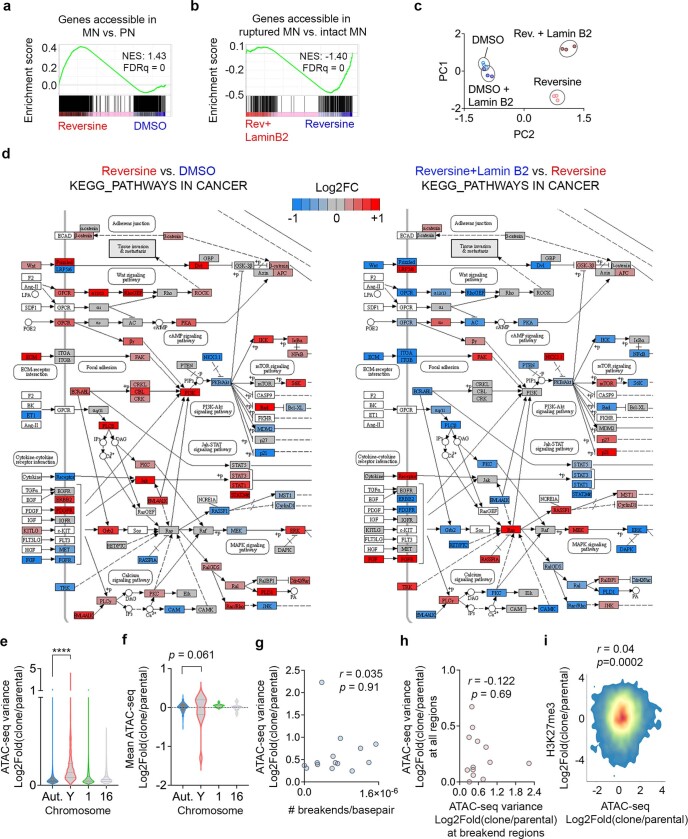
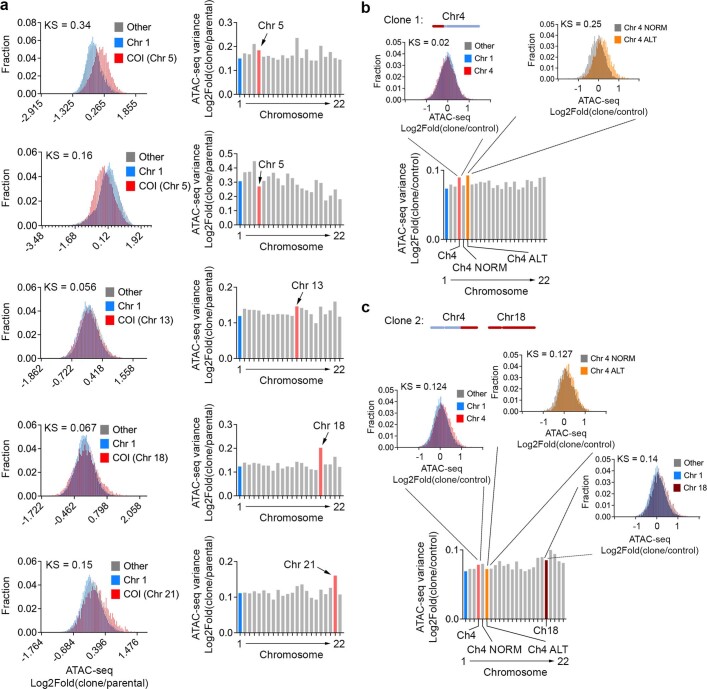
Chromosomal instability (CIN) and epigenetic alterations are characteristics of advanced and metastatic cancers1-4, but whether they are mechanistically linked is unknown. Here we show that missegregation of mitotic chromosomes, their sequestration in micronuclei5,6 and subsequent rupture of the micronuclear envelope7 profoundly disrupt normal histone post-translational modifications (PTMs), a phenomenon conserved across humans and mice, as well as in cancer and non-transformed cells. Some of the changes in histone PTMs occur because of the rupture of the micronuclear envelope, whereas others are inherited from mitotic abnormalities before the micronucleus is formed. Using orthogonal approaches, we demonstrate that micronuclei exhibit extensive differences in chromatin accessibility, with a strong positional bias between promoters and distal or intergenic regions, in line with observed redistributions of histone PTMs. Inducing CIN causes widespread epigenetic dysregulation, and chromosomes that transit in micronuclei experience heritable abnormalities in their accessibility long after they have been reincorporated into the primary nucleus. Thus, as well as altering genomic copy number, CIN promotes epigenetic reprogramming and heterogeneity in cancer.
© 2023. The Author(s).
Conflict of interest statement
S.F.B. owns equity in, receives compensation from, serves as a consultant to, and serves on the scientific advisory board and board of directors of Volastra Therapeutics. V.B. is a co-founder, consultant, member of the scientific advisory board and has equity interest in Boundless Bio and Abterra. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies. P.S.M. is a co-founder of Boundless Bio. He has equity in the company and chairs the scientific advisory board, for which he is compensated. B.W. reports a research grant from Repare Therapeutics, outside the submitted work. S.P.S. is a shareholder in Imagia Canexia Health and is a consultant with Astra Zeneca Inc, outside the submitted work, R.P.K. is a co-founder of and consultant for Econic Biosciences. The other authors declare no competing interests.
Figures
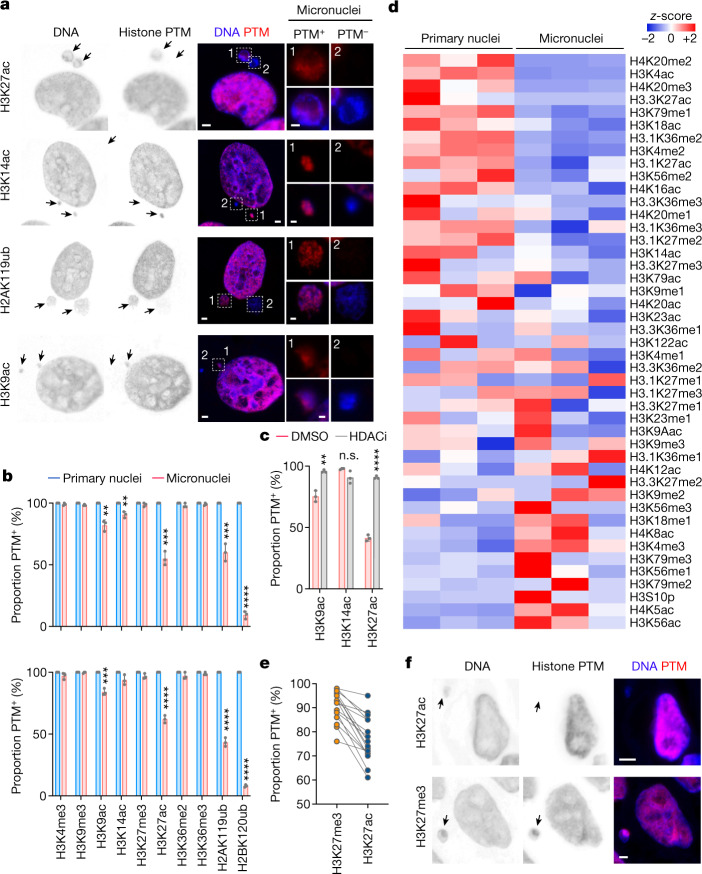
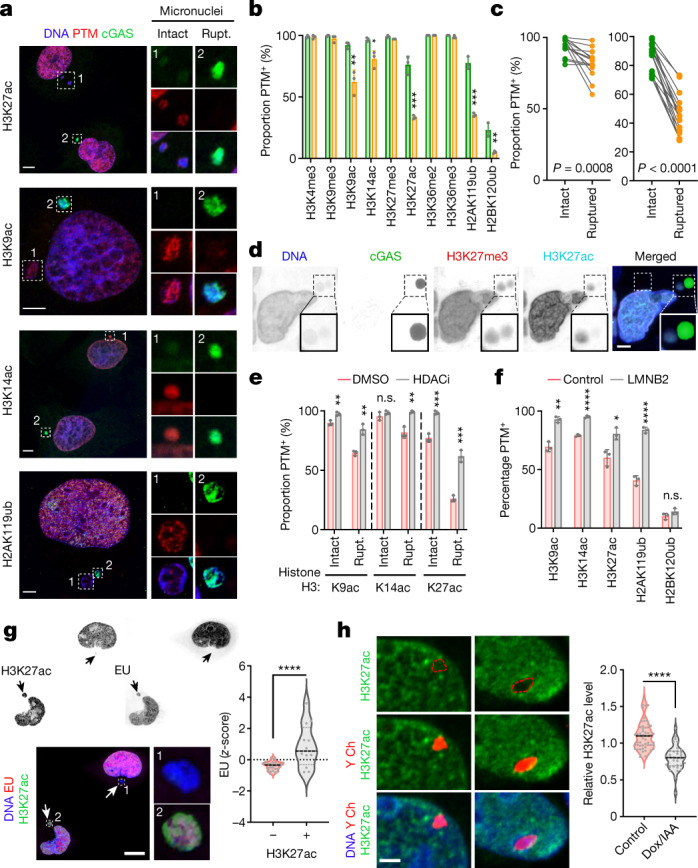
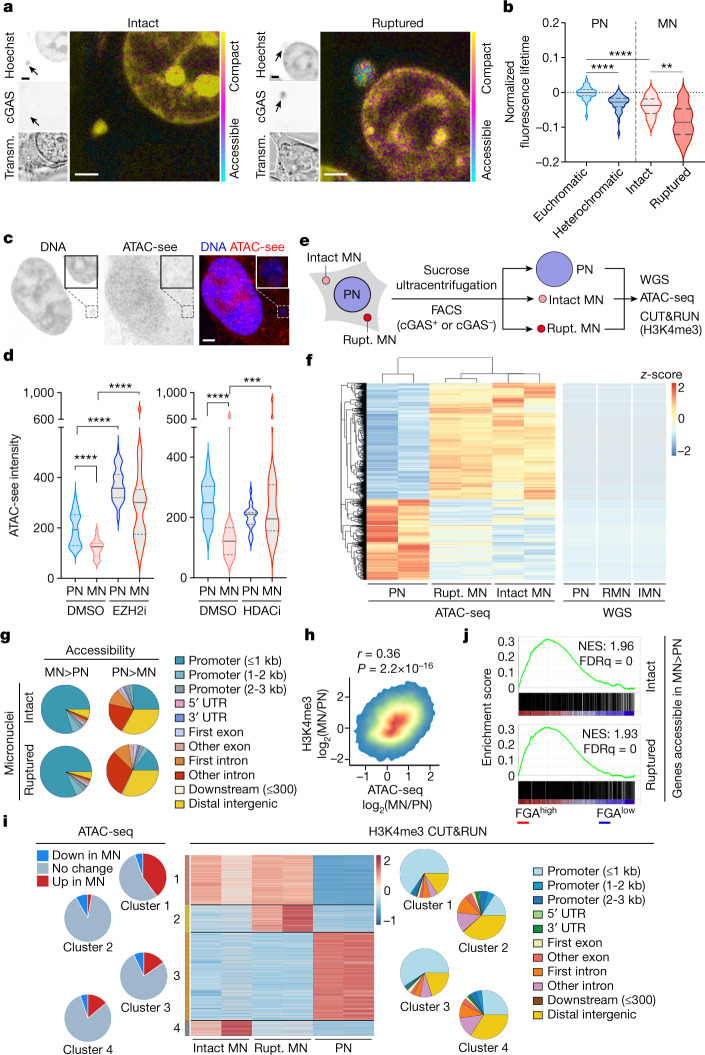
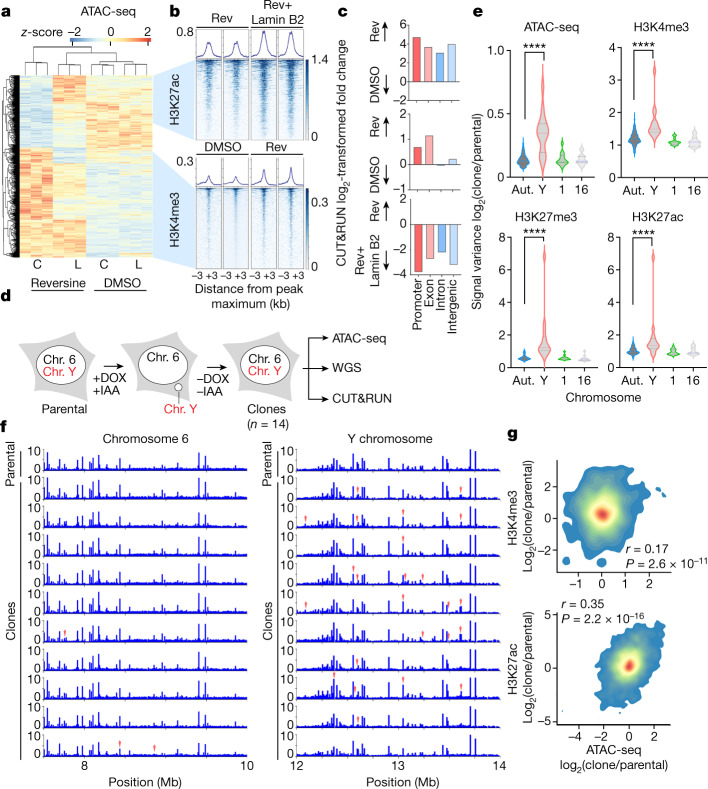
References
-
- Kato H, Sandberg AA. Chromosome pulverization in human cells with micronuclei. J. Natl Cancer Inst. 1968;40:165–179. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DP5 OD026395/OD/NIH HHS/United States
- P50 CA192937/CA/NCI NIH HHS/United States
- R37 CA261183/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R35 GM138386/GM/NIGMS NIH HHS/United States
- R01 GM114362/GM/NIGMS NIH HHS/United States
- R01 CA256188/CA/NCI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- T32 CA009207/CA/NCI NIH HHS/United States
- R01 CA270102/CA/NCI NIH HHS/United States
- U24 CA264379/CA/NCI NIH HHS/United States
- P50 CA247749/CA/NCI NIH HHS/United States
- S10 OD030286/OD/NIH HHS/United States
- P30 CA016359/CA/NCI NIH HHS/United States
- T32 GM007739/GM/NIGMS NIH HHS/United States
- R00 CA212290/CA/NCI NIH HHS/United States
- L32 MD017781/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
