

Ultraviolet radiation shapes dendritic cell leukaemia transformation in the skin
- PMID: 37286599
- PMCID: PMC10284703
- DOI: 10.1038/s41586-023-06156-8
Ultraviolet radiation shapes dendritic cell leukaemia transformation in the skin
Abstract
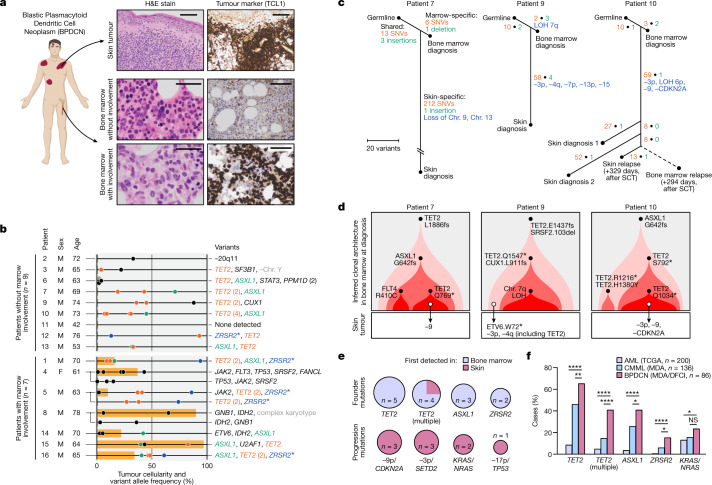
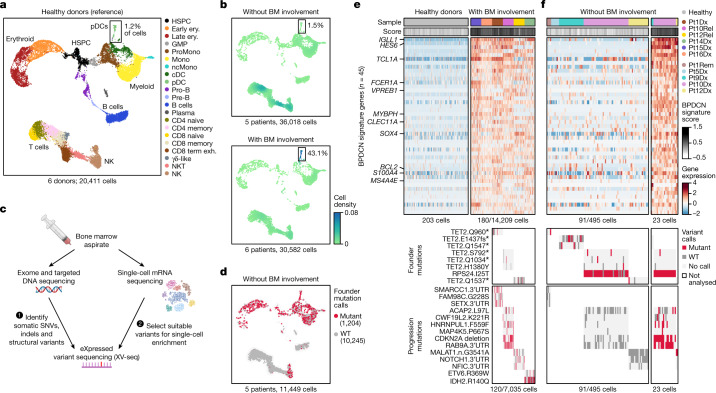
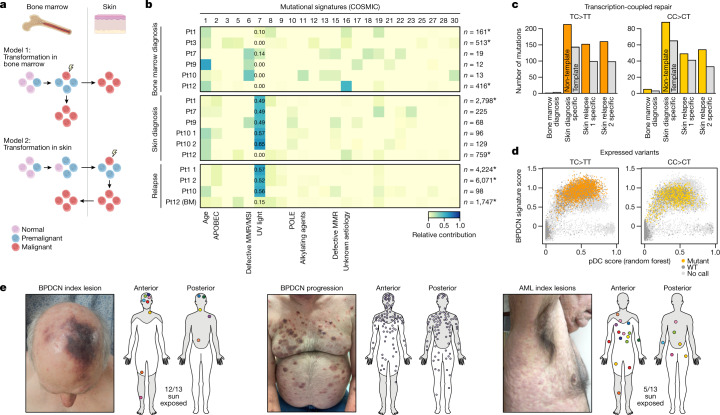
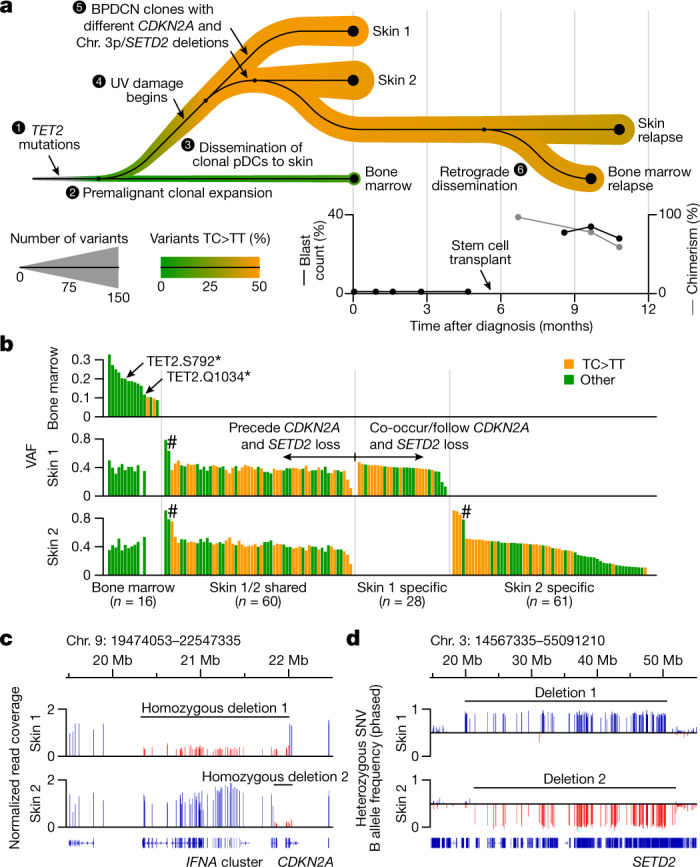
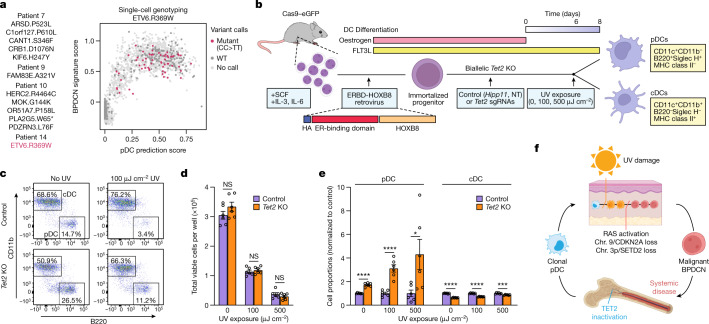
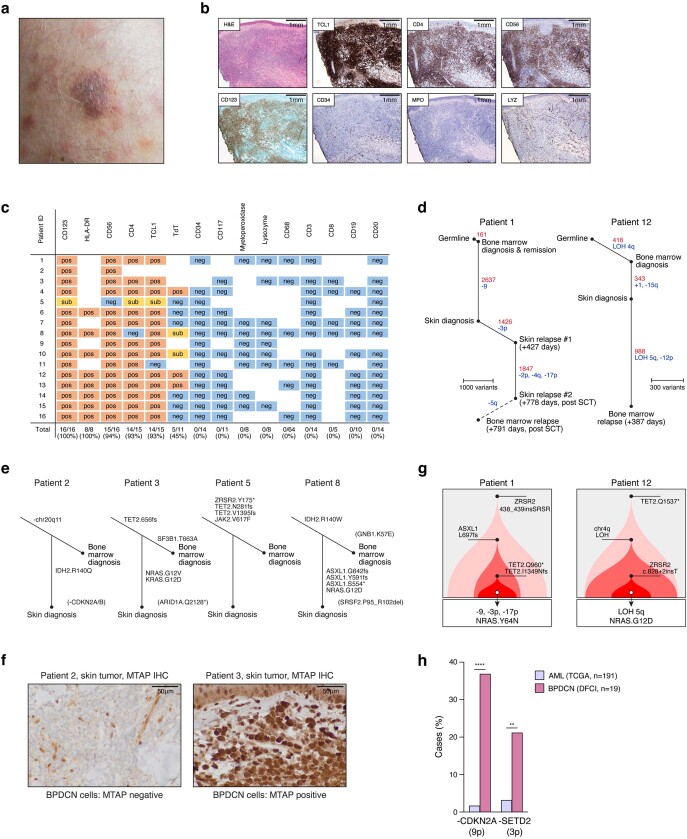
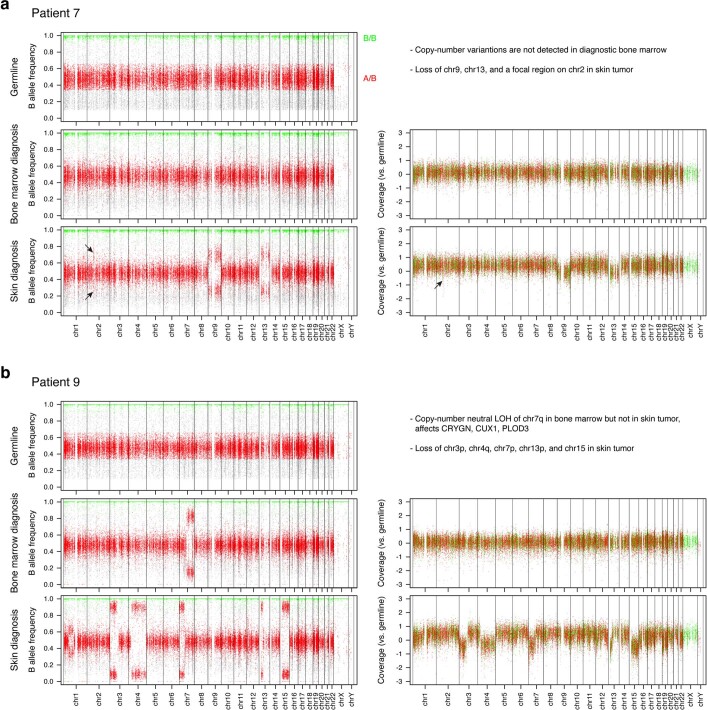
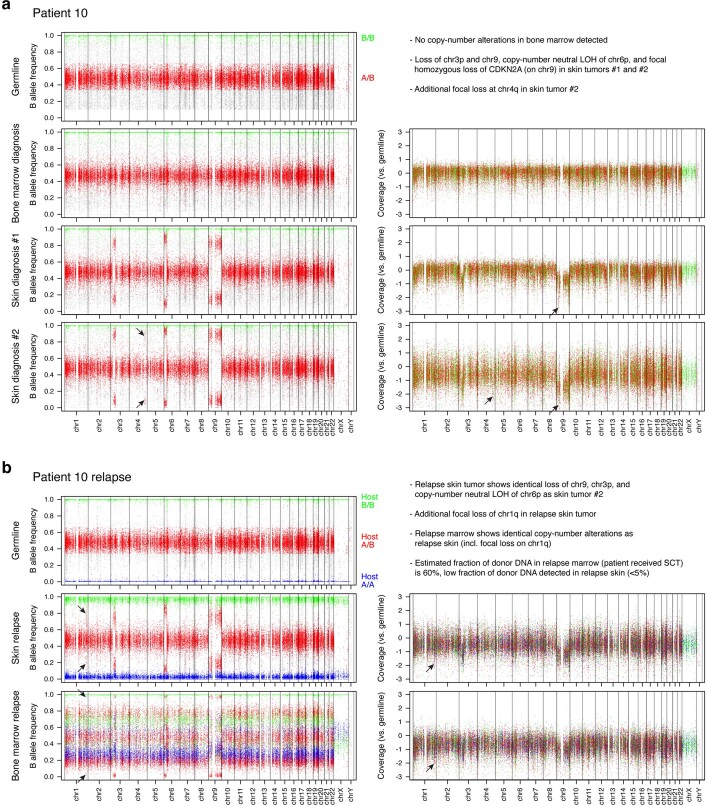
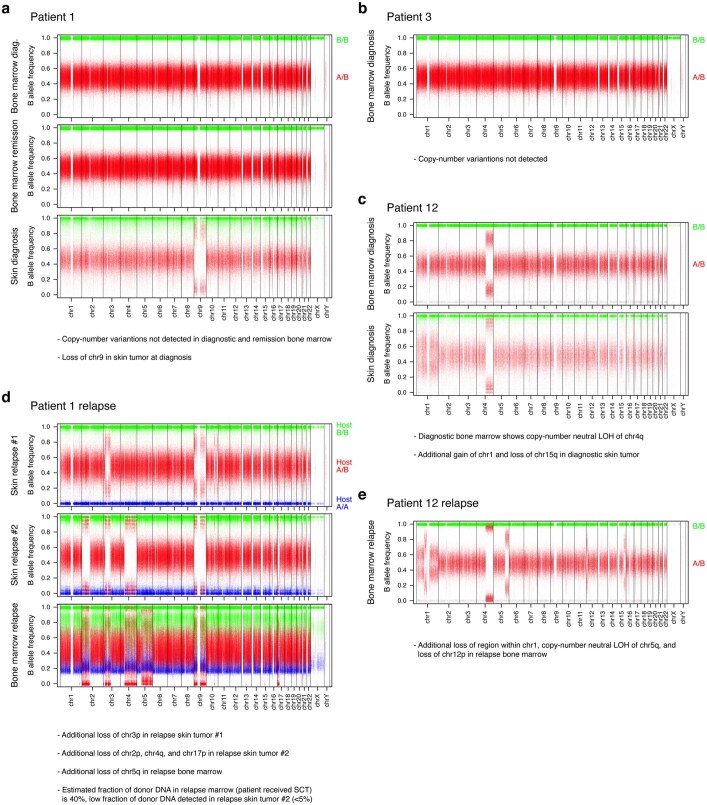
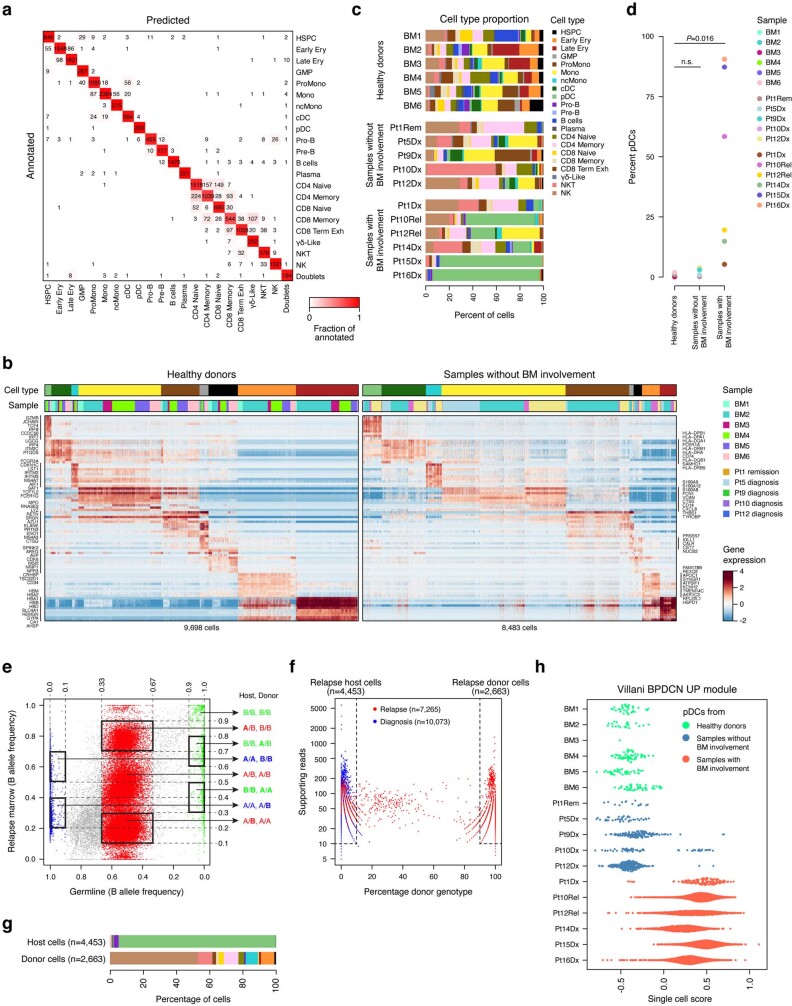
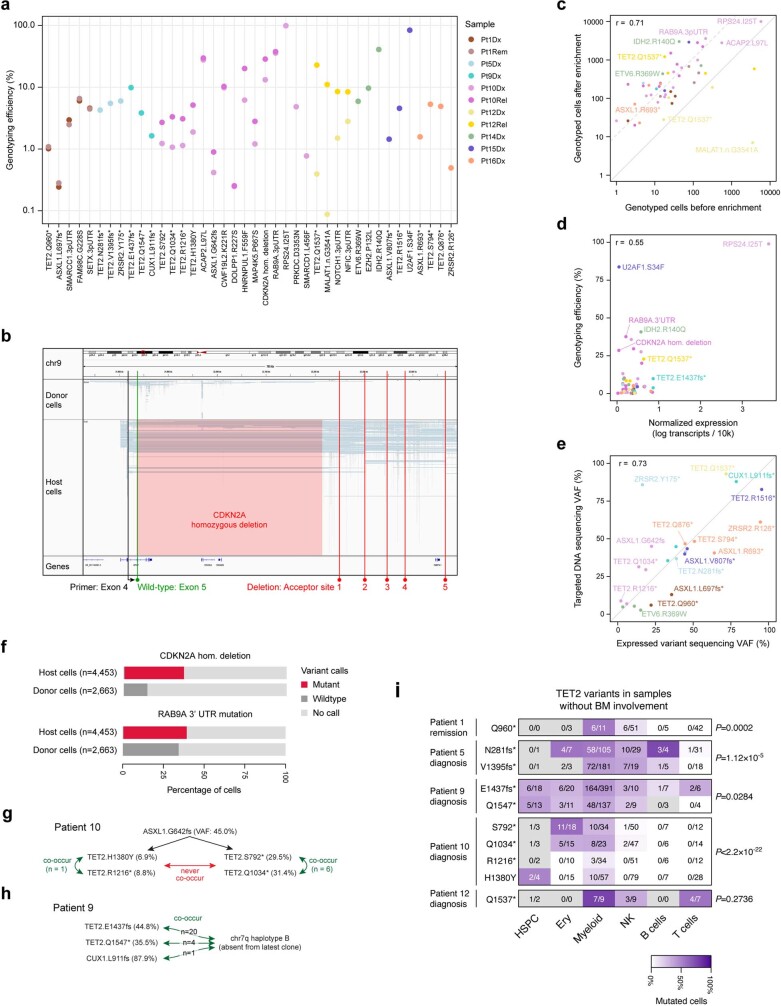
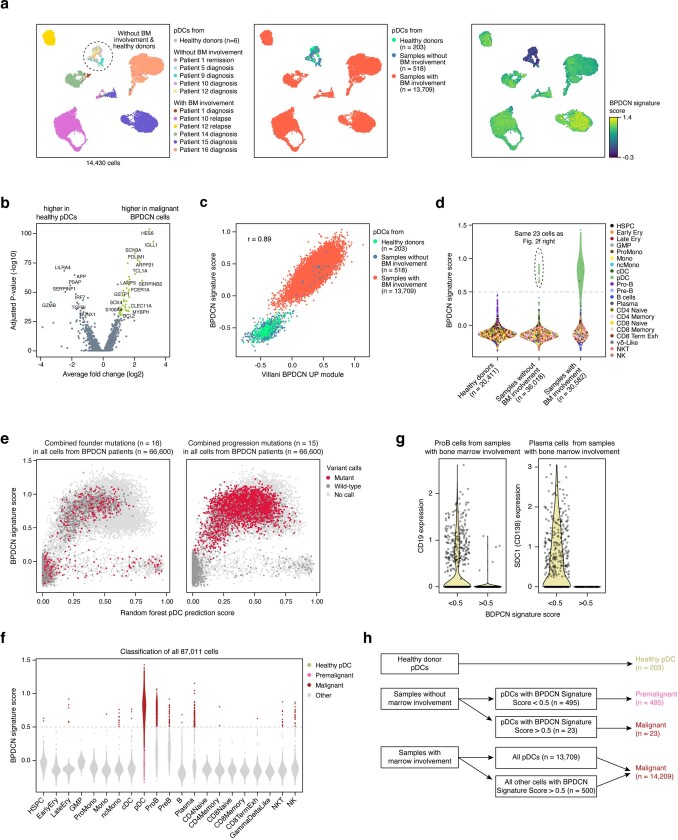
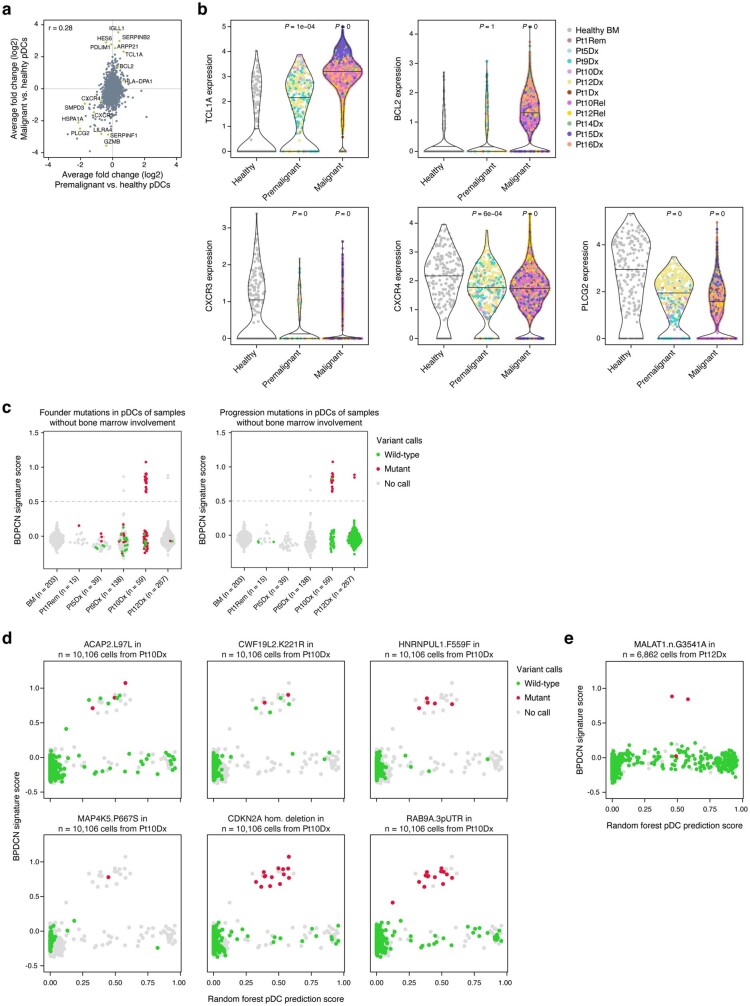
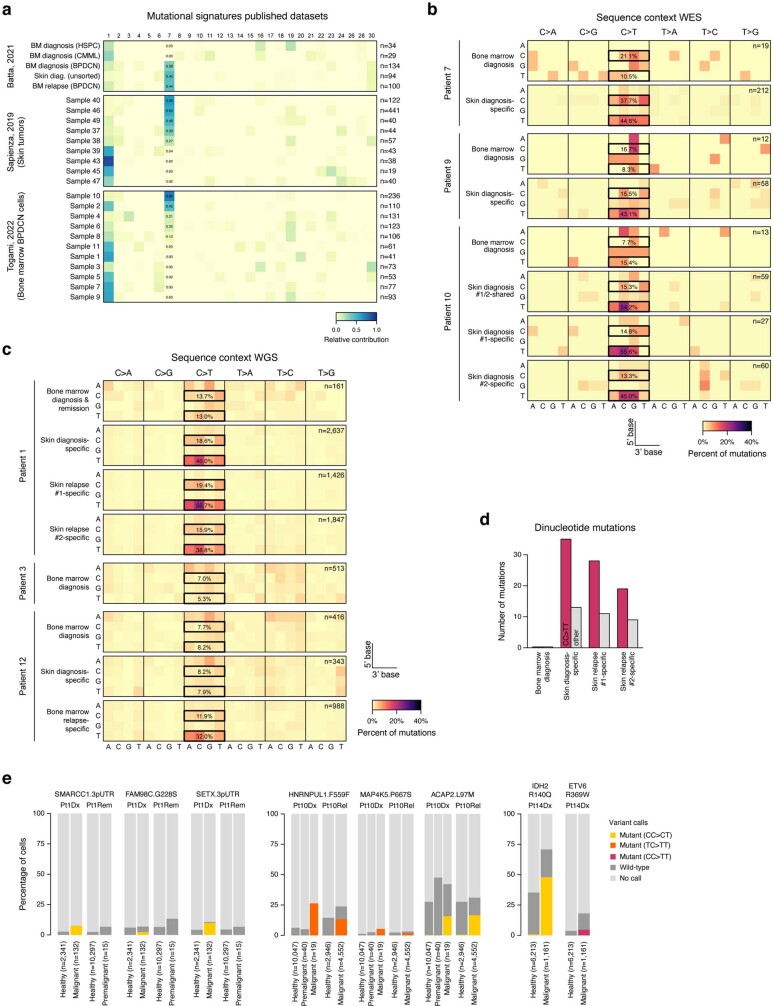
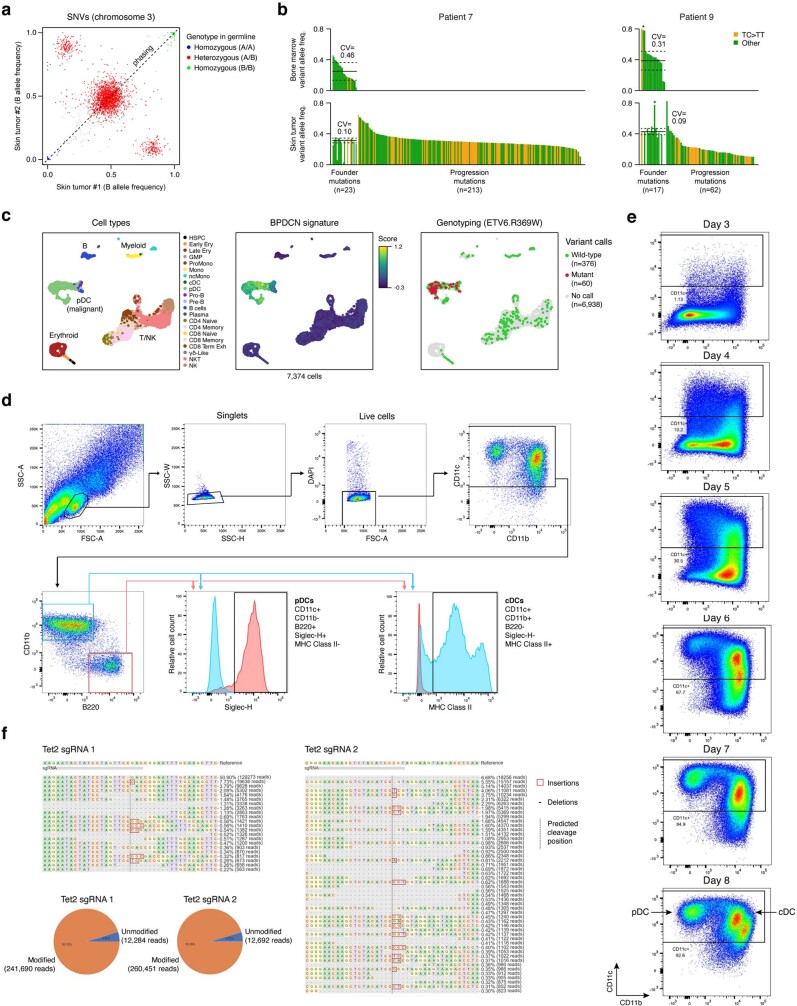
Tumours most often arise from progression of precursor clones within a single anatomical niche. In the bone marrow, clonal progenitors can undergo malignant transformation to acute leukaemia, or differentiate into immune cells that contribute to disease pathology in peripheral tissues1-4. Outside the marrow, these clones are potentially exposed to a variety of tissue-specific mutational processes, although the consequences of this are unclear. Here we investigate the development of blastic plasmacytoid dendritic cell neoplasm (BPDCN)-an unusual form of acute leukaemia that often presents with malignant cells isolated to the skin5. Using tumour phylogenomics and single-cell transcriptomics with genotyping, we find that BPDCN arises from clonal (premalignant) haematopoietic precursors in the bone marrow. We observe that BPDCN skin tumours first develop at sun-exposed anatomical sites and are distinguished by clonally expanded mutations induced by ultraviolet (UV) radiation. A reconstruction of tumour phylogenies reveals that UV damage can precede the acquisition of alterations associated with malignant transformation, implicating sun exposure of plasmacytoid dendritic cells or committed precursors during BPDCN pathogenesis. Functionally, we find that loss-of-function mutations in Tet2, the most common premalignant alteration in BPDCN, confer resistance to UV-induced cell death in plasmacytoid, but not conventional, dendritic cells, suggesting a context-dependent tumour-suppressive role for TET2. These findings demonstrate how tissue-specific environmental exposures at distant anatomical sites can shape the evolution of premalignant clones to disseminated cancer.
© 2023. The Author(s).
Conflict of interest statement
B.E.B. discloses financial interests in Fulcrum Therapeutics, Chroma Medicine, HiFiBio, Arsenal Biosciences, Cell Signaling Technologies and Design Pharmaceuticals. A.A.L. received research funding from Abbvie and Stemline therapeutics, and consulting fees from Cimeio Therapeutics, IDRx, Jnana Therapeutics, N-of-One and Qiagen. P.v.G. received consulting fees from ManaT Bio and Immunitas.
Figures
Comment in
-
Ultraviolet light shapes the evolution of precancerous cells.Nature. 2023 Jun;618(7966):681-683. doi: 10.1038/d41586-023-01815-2. Nature. 2023. PMID: 37286674 No abstract available.
-
Tissue-Specific Exposures Dictate the Evolution of Dendritic Cell Leukemia.Cancer Discov. 2023 Aug 4;13(8):1760. doi: 10.1158/2159-8290.CD-RW2023-099. Cancer Discov. 2023. PMID: 37350647
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
