

Heritable transcriptional defects from aberrations of nuclear architecture
- PMID: 37286600
- PMCID: PMC10322708
- DOI: 10.1038/s41586-023-06157-7
Heritable transcriptional defects from aberrations of nuclear architecture
Abstract
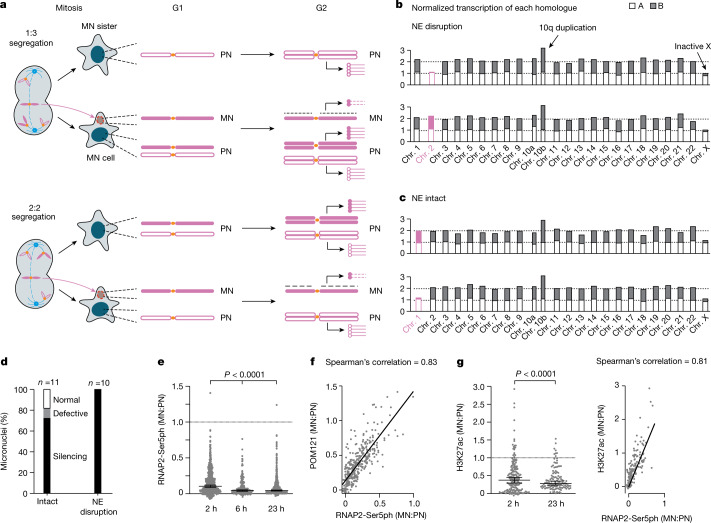
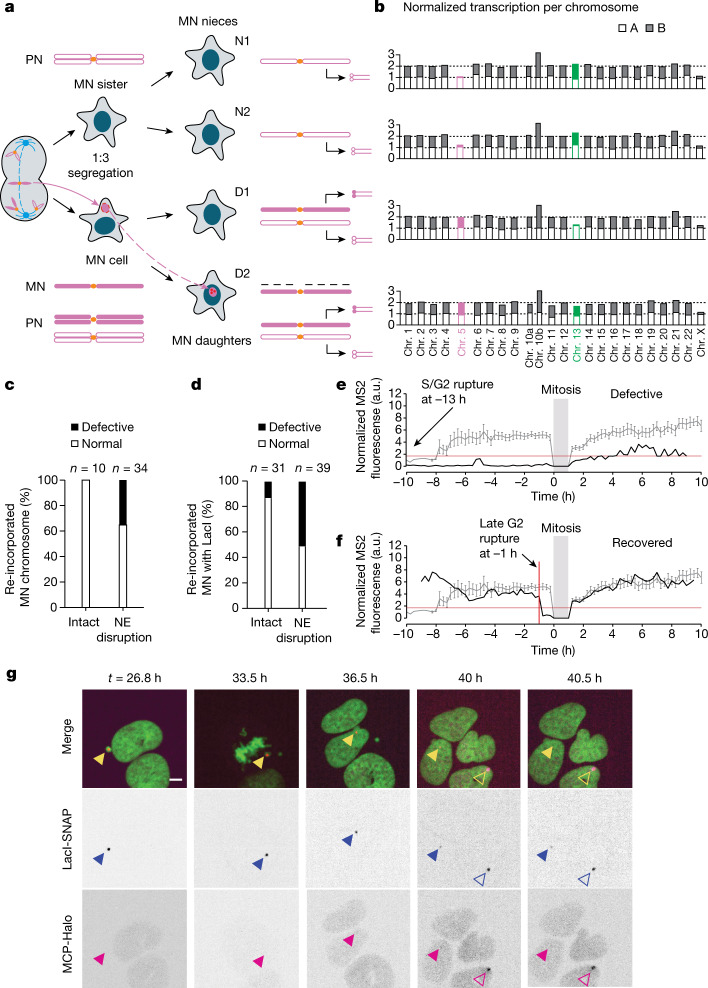
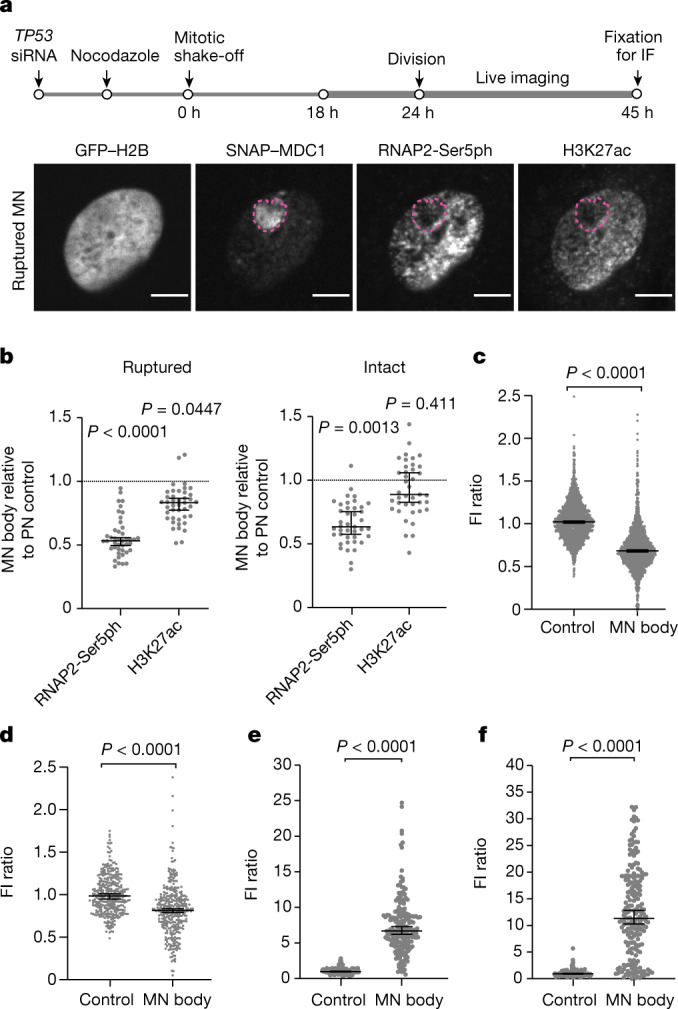
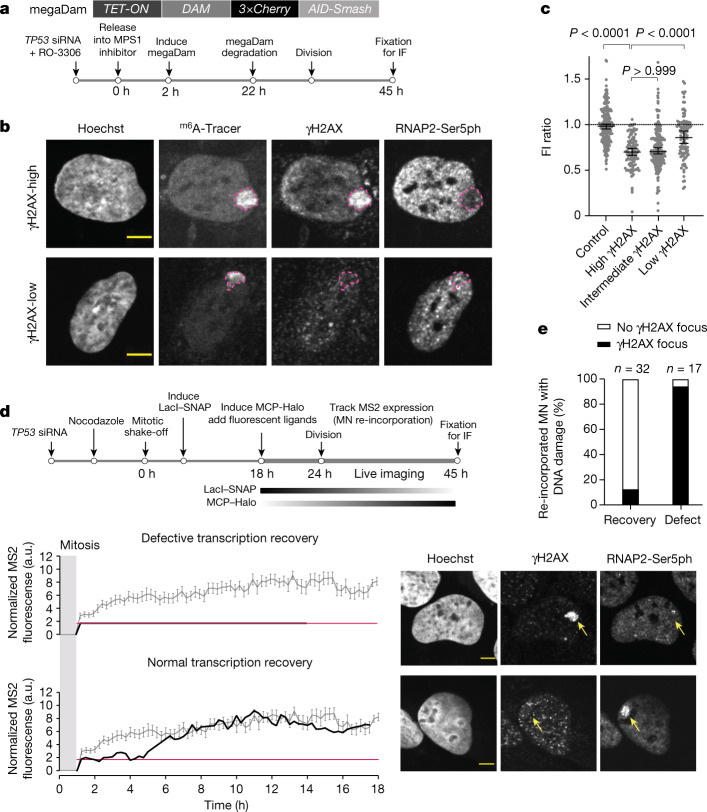
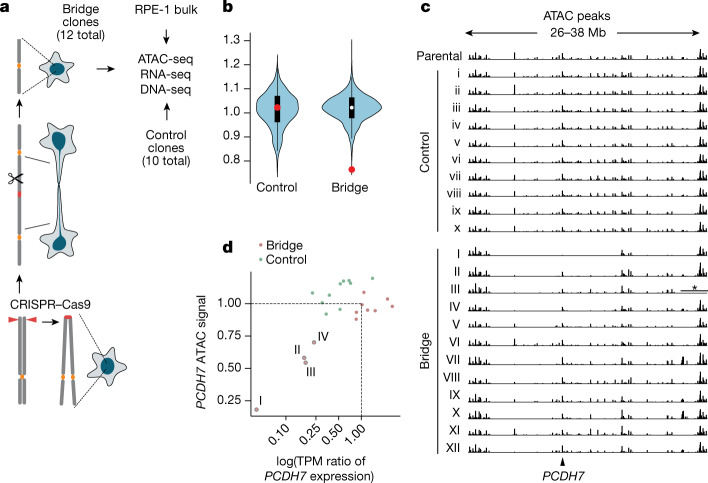
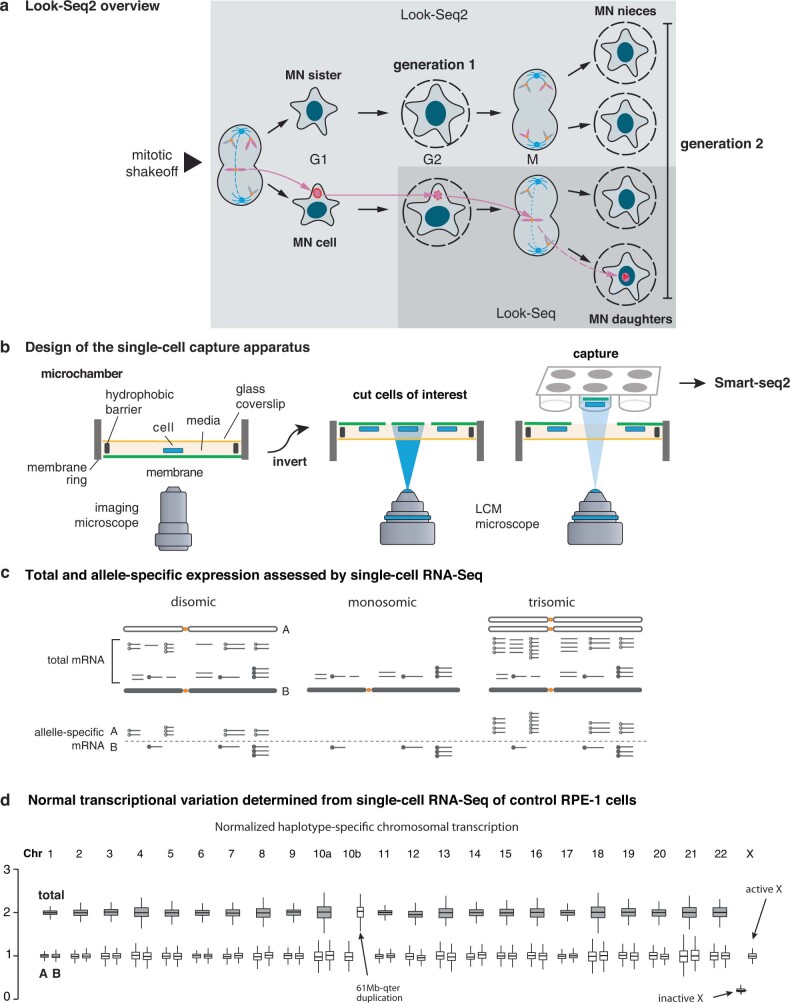
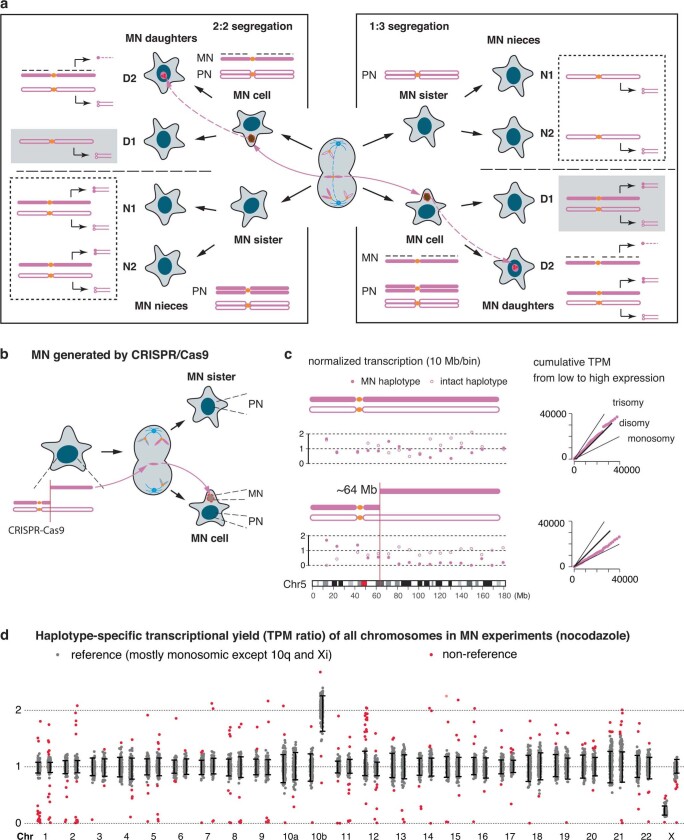
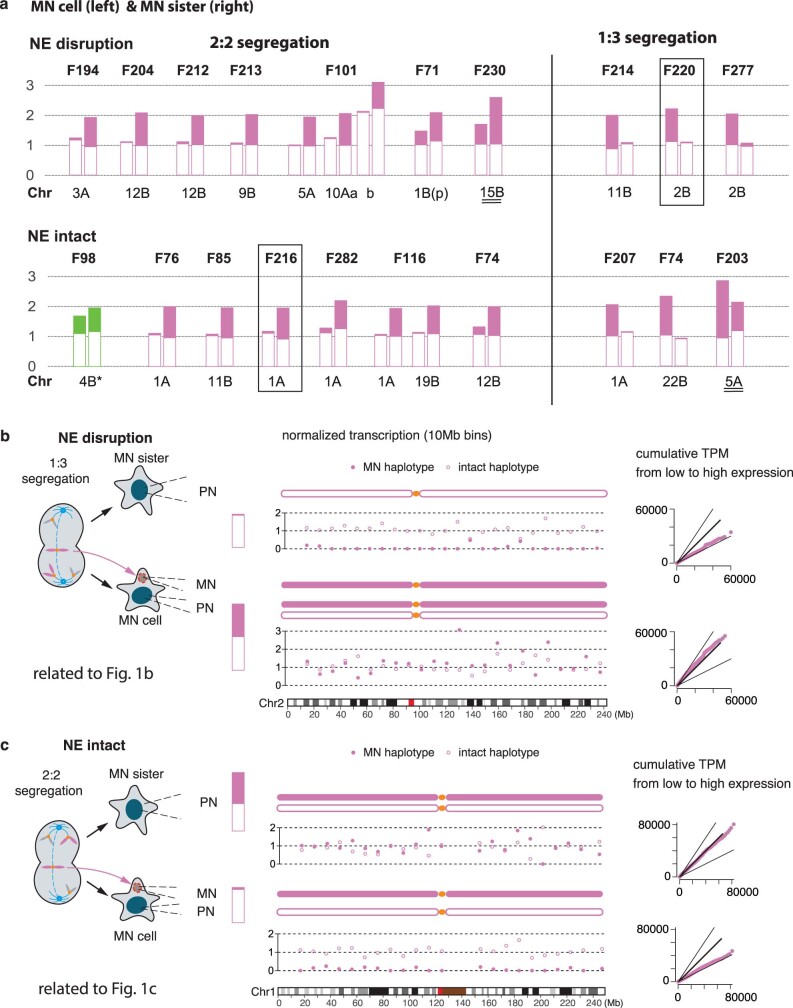
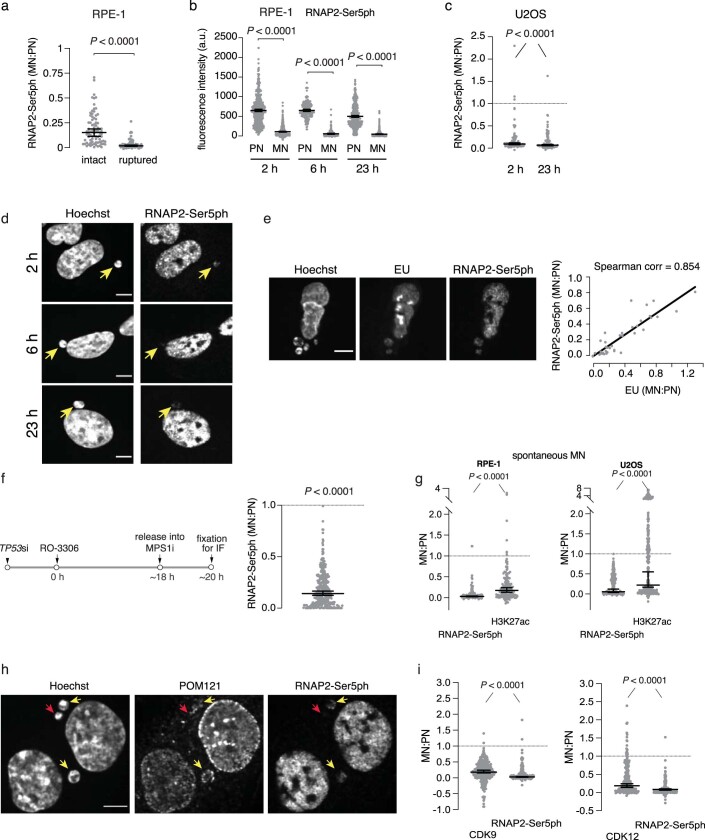
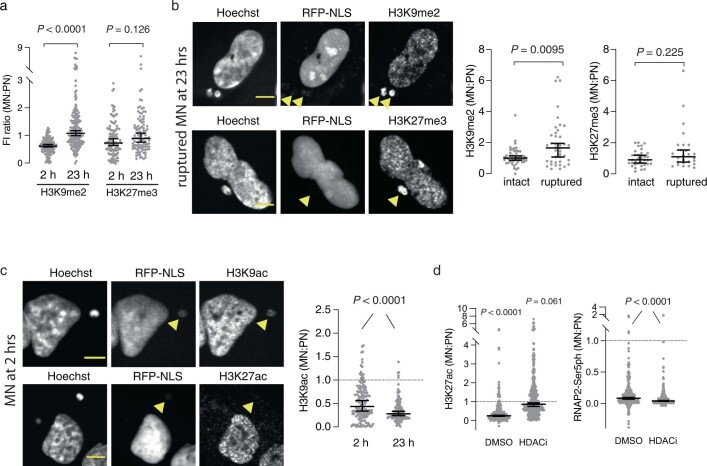
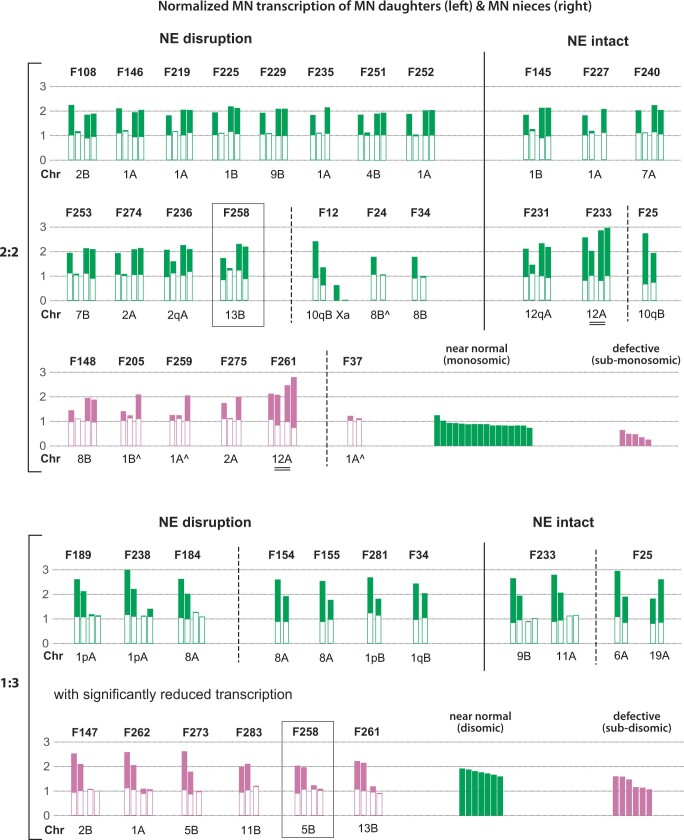
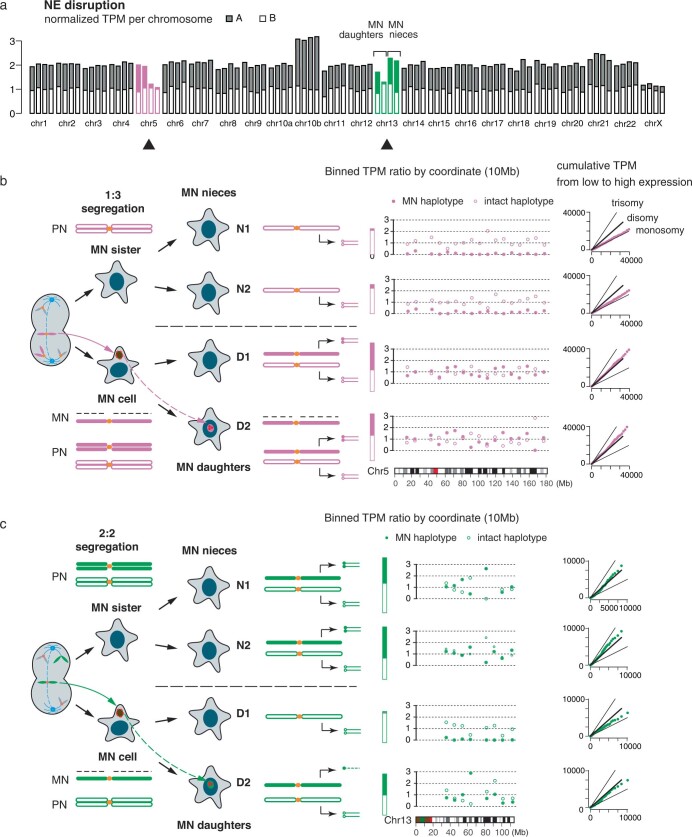
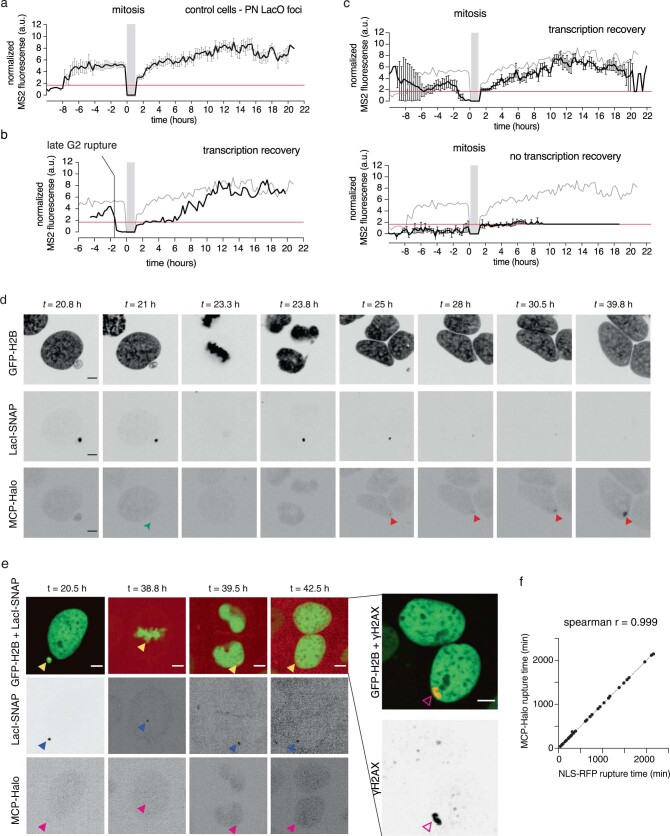
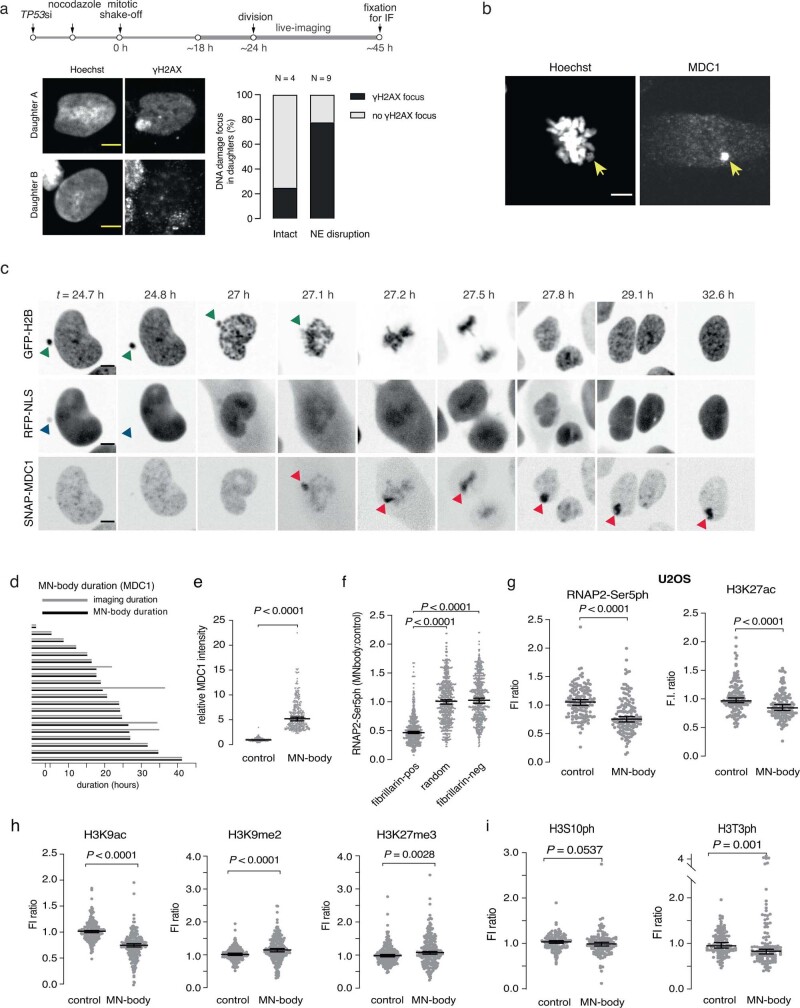
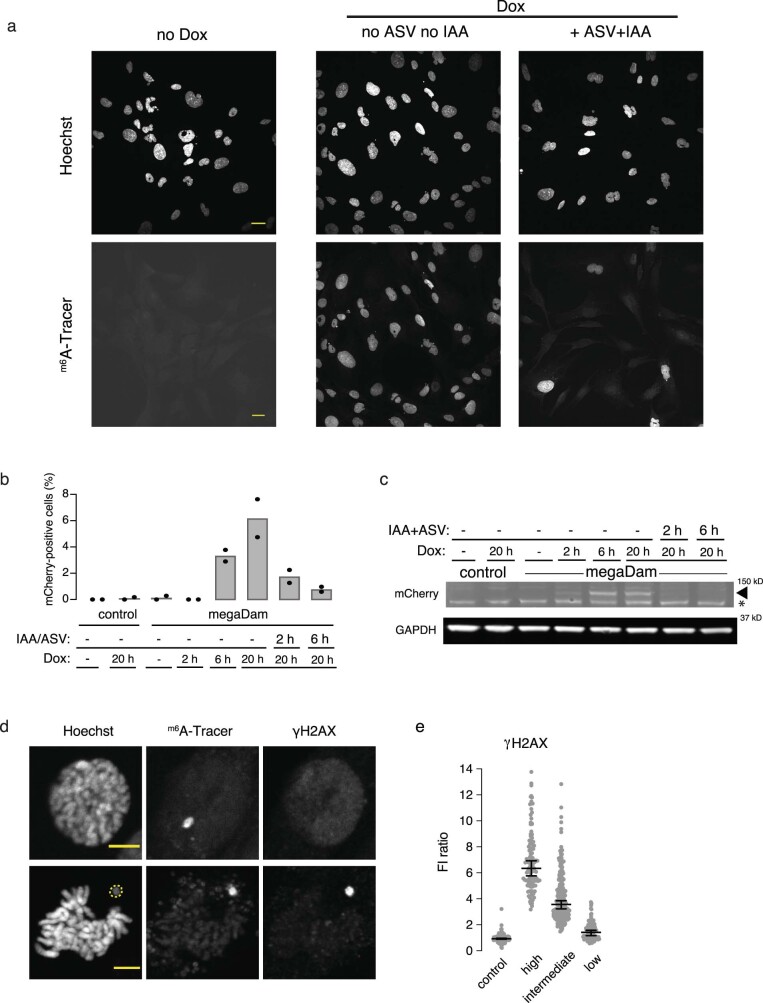
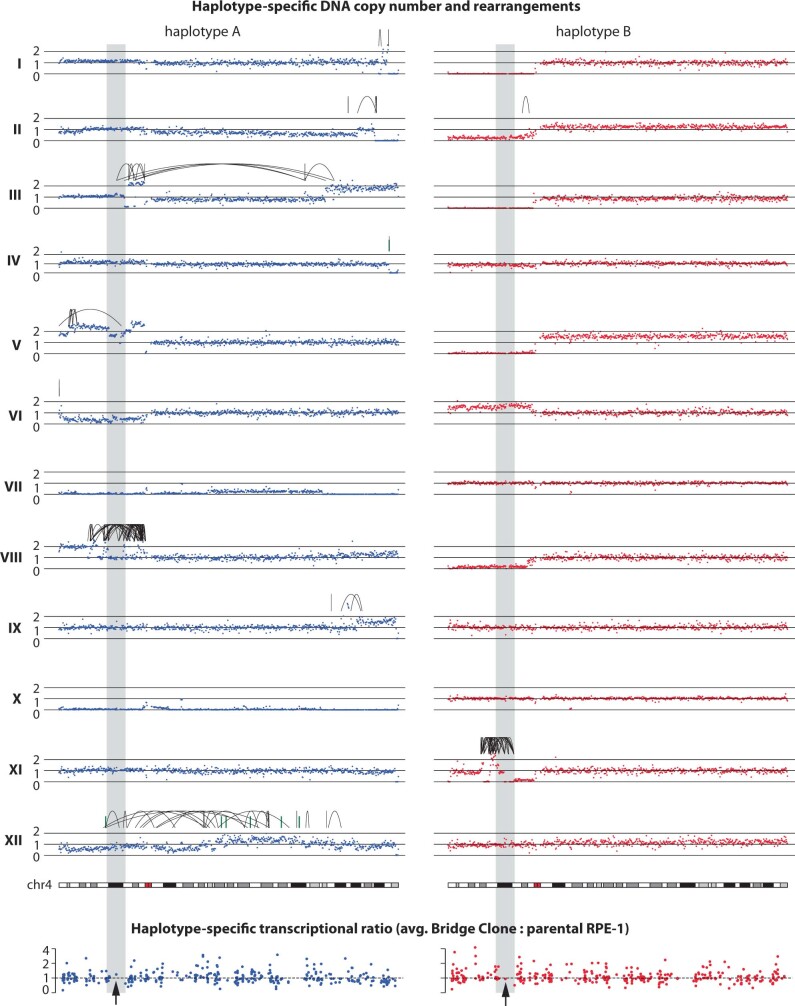
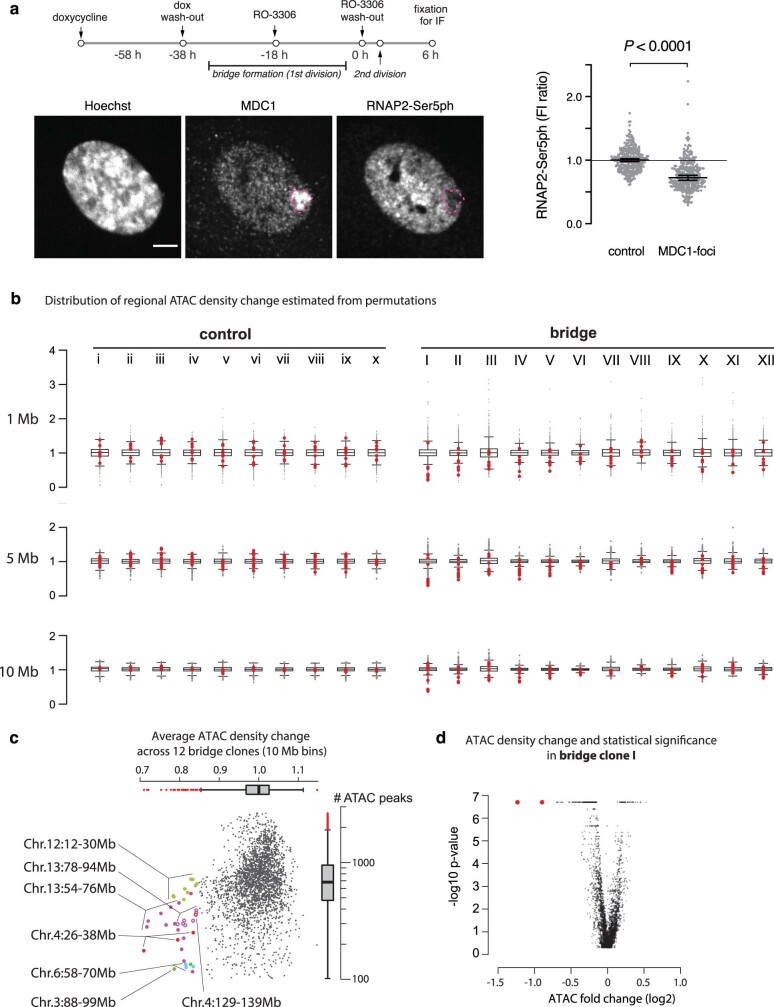
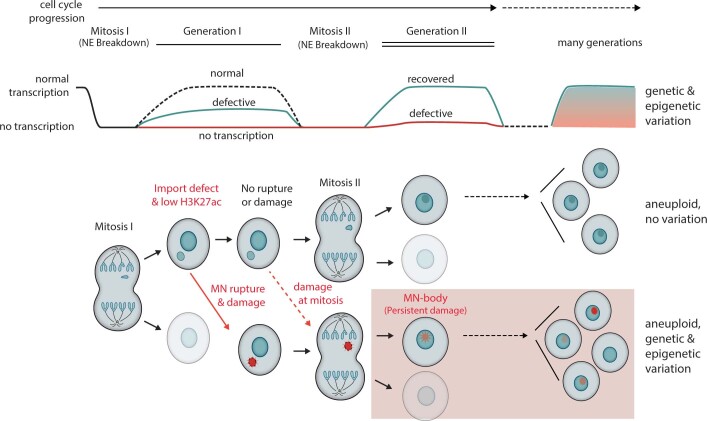
Transcriptional heterogeneity due to plasticity of the epigenetic state of chromatin contributes to tumour evolution, metastasis and drug resistance1-3. However, the mechanisms that cause this epigenetic variation are incompletely understood. Here we identify micronuclei and chromosome bridges, aberrations in the nucleus common in cancer4,5, as sources of heritable transcriptional suppression. Using a combination of approaches, including long-term live-cell imaging and same-cell single-cell RNA sequencing (Look-Seq2), we identified reductions in gene expression in chromosomes from micronuclei. With heterogeneous penetrance, these changes in gene expression can be heritable even after the chromosome from the micronucleus has been re-incorporated into a normal daughter cell nucleus. Concomitantly, micronuclear chromosomes acquire aberrant epigenetic chromatin marks. These defects may persist as variably reduced chromatin accessibility and reduced gene expression after clonal expansion from single cells. Persistent transcriptional repression is strongly associated with, and may be explained by, markedly long-lived DNA damage. Epigenetic alterations in transcription may therefore be inherently coupled to chromosomal instability and aberrations in nuclear architecture.
© 2023. The Author(s).
Conflict of interest statement
J.D.B. holds patents related to ATAC-seq and is a scientific advisory board member of Camp4 and seqWell. C.-Z.Z. is a scientific adviser for Pillar BioSciences. D.P. is a member of the Volastra Therapeutics scientific advisory board. Dana-Farber Cancer Institute is in the process of applying for a patent application (PCT/US20 19/023696, September 26, 2019) covering “Systems and methods for capturing cells” that lists S.P. as inventor. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
