

Pluripotency-independent induction of human trophoblast stem cells from fibroblasts
- PMID: 37291192
- PMCID: PMC10250329
- DOI: 10.1038/s41467-023-39104-1
Pluripotency-independent induction of human trophoblast stem cells from fibroblasts
Abstract
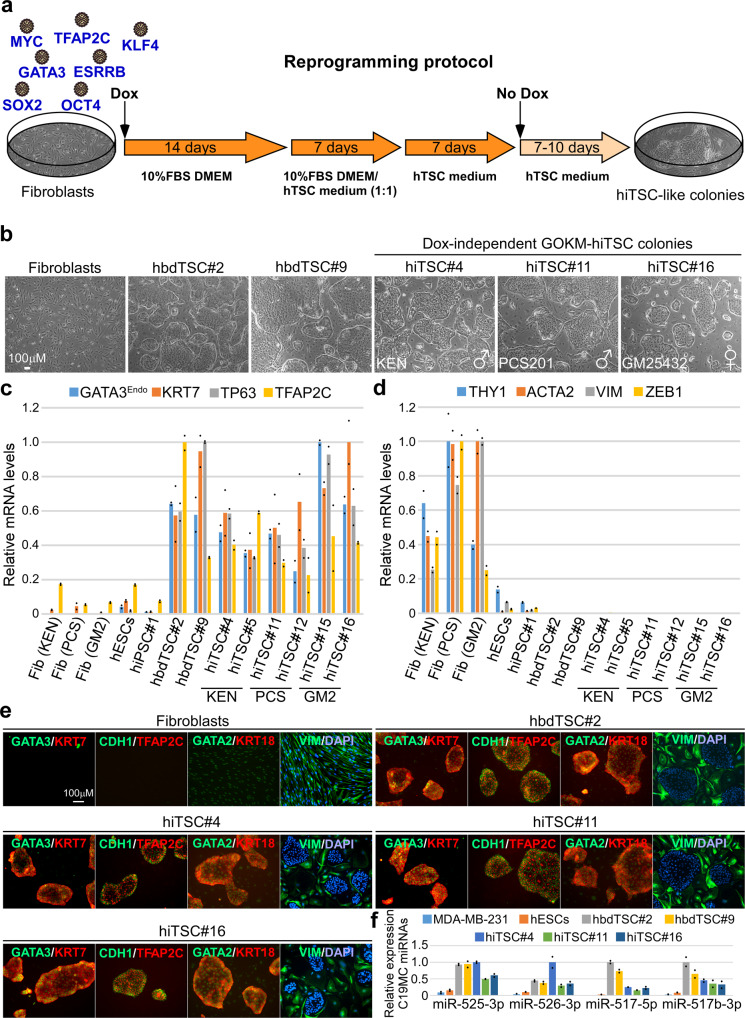
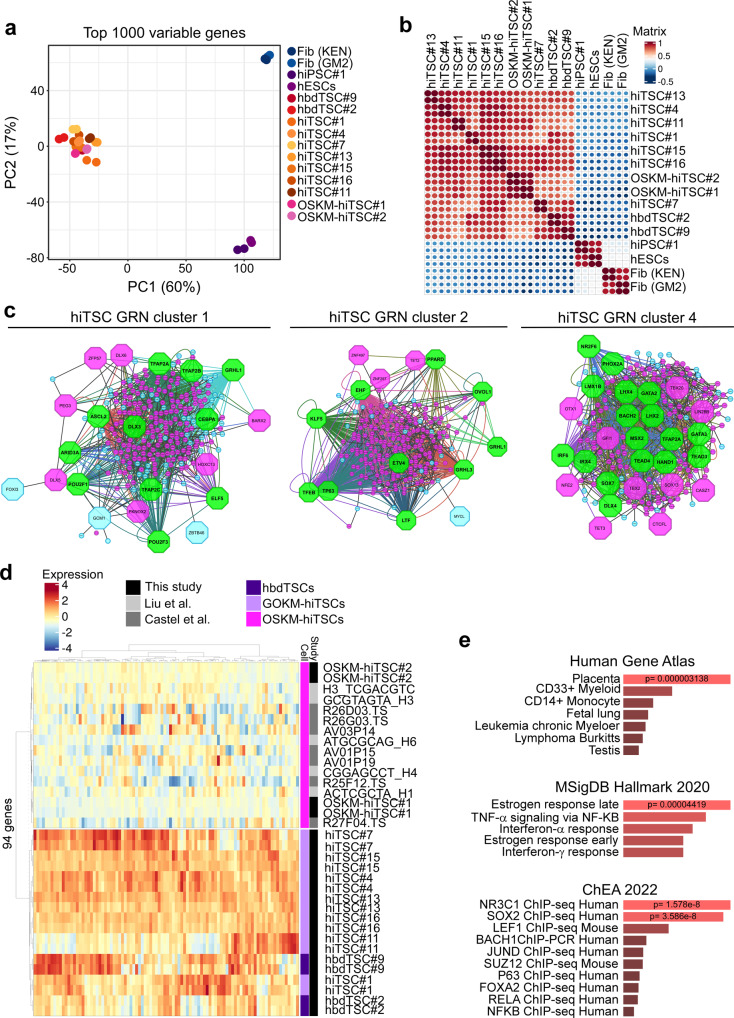
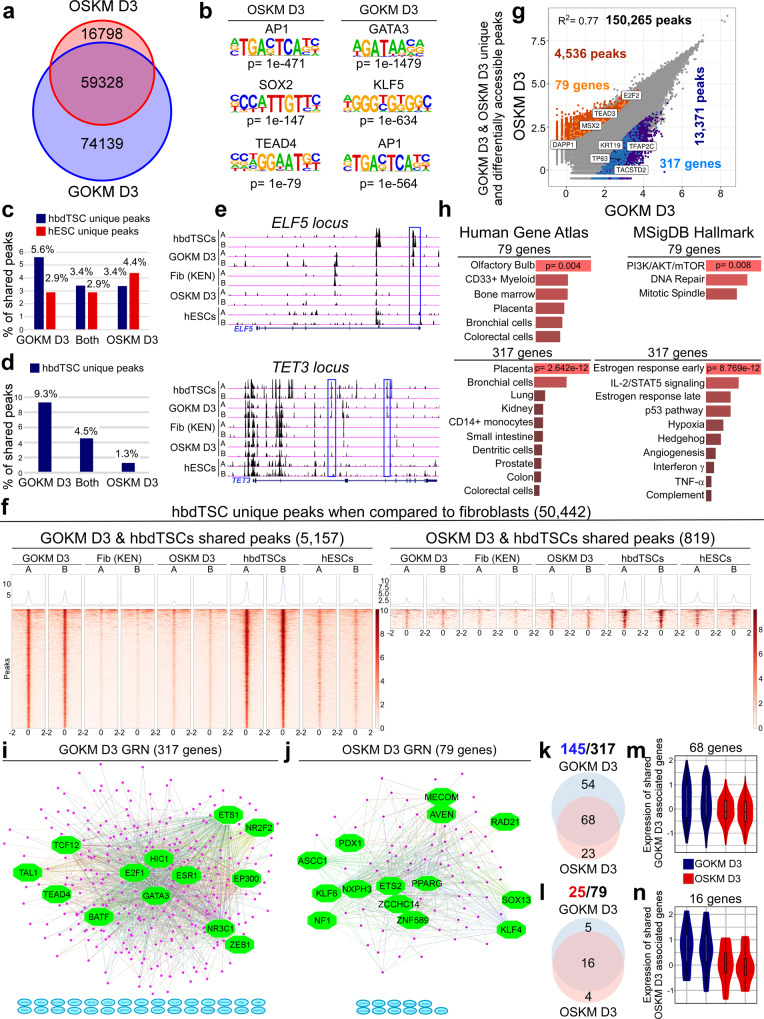
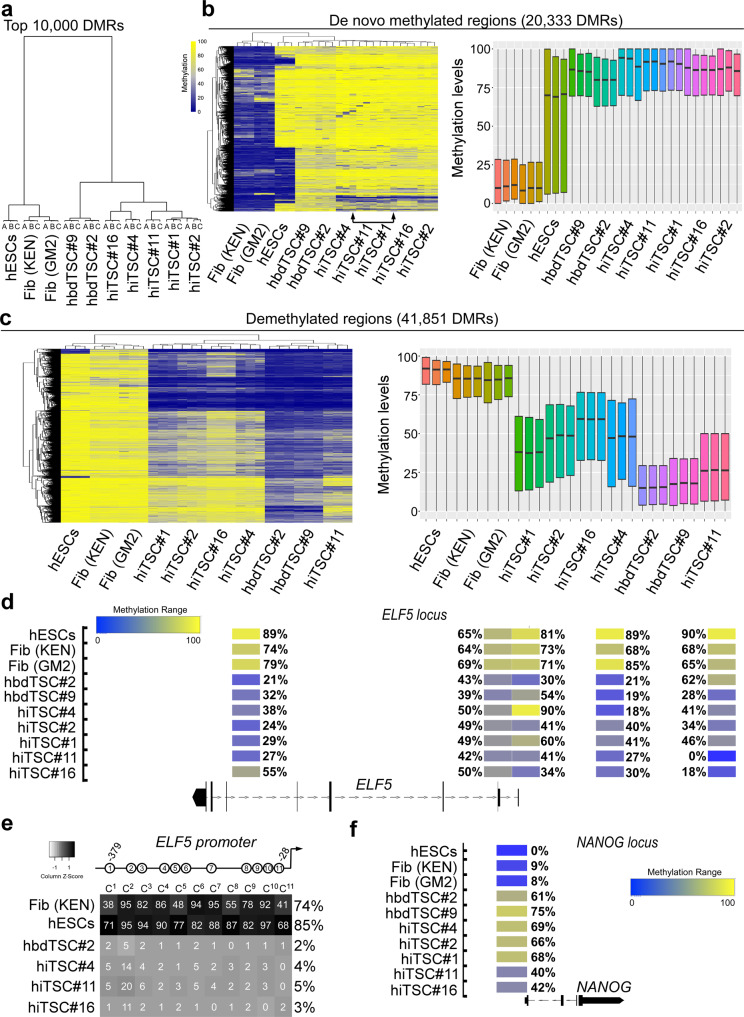
Human trophoblast stem cells (hTSCs) can be derived from embryonic stem cells (hESCs) or be induced from somatic cells by OCT4, SOX2, KLF4 and MYC (OSKM). Here we explore whether the hTSC state can be induced independently of pluripotency, and what are the mechanisms underlying its acquisition. We identify GATA3, OCT4, KLF4 and MYC (GOKM) as a combination of factors that can generate functional hiTSCs from fibroblasts. Transcriptomic analysis of stable GOKM- and OSKM-hiTSCs reveals 94 hTSC-specific genes that are aberrant specifically in OSKM-derived hiTSCs. Through time-course-RNA-seq analysis, H3K4me2 deposition and chromatin accessibility, we demonstrate that GOKM exert greater chromatin opening activity than OSKM. While GOKM primarily target hTSC-specific loci, OSKM mainly induce the hTSC state via targeting hESC and hTSC shared loci. Finally, we show that GOKM efficiently generate hiTSCs from fibroblasts that harbor knockout for pluripotency genes, further emphasizing that pluripotency is dispensable for hTSC state acquisition.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
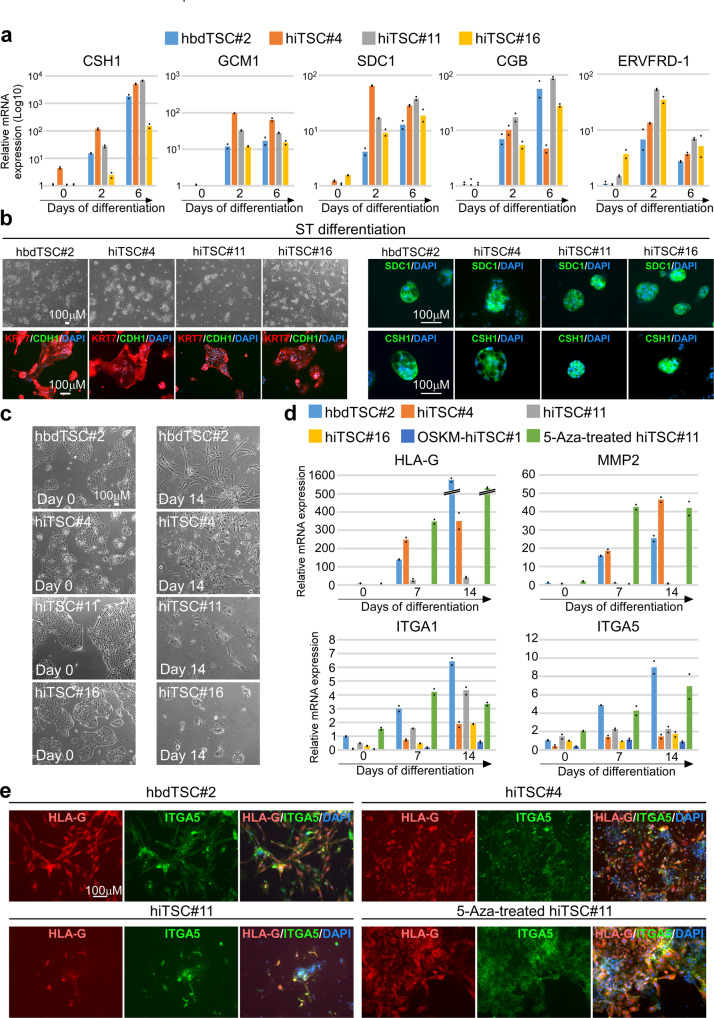
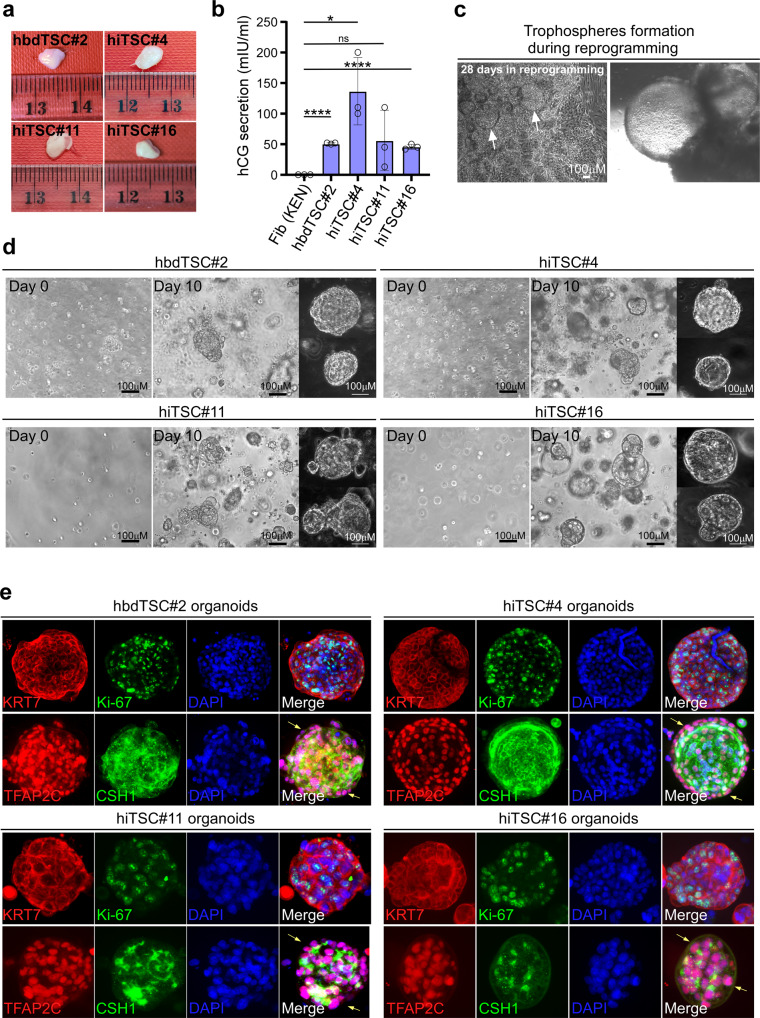
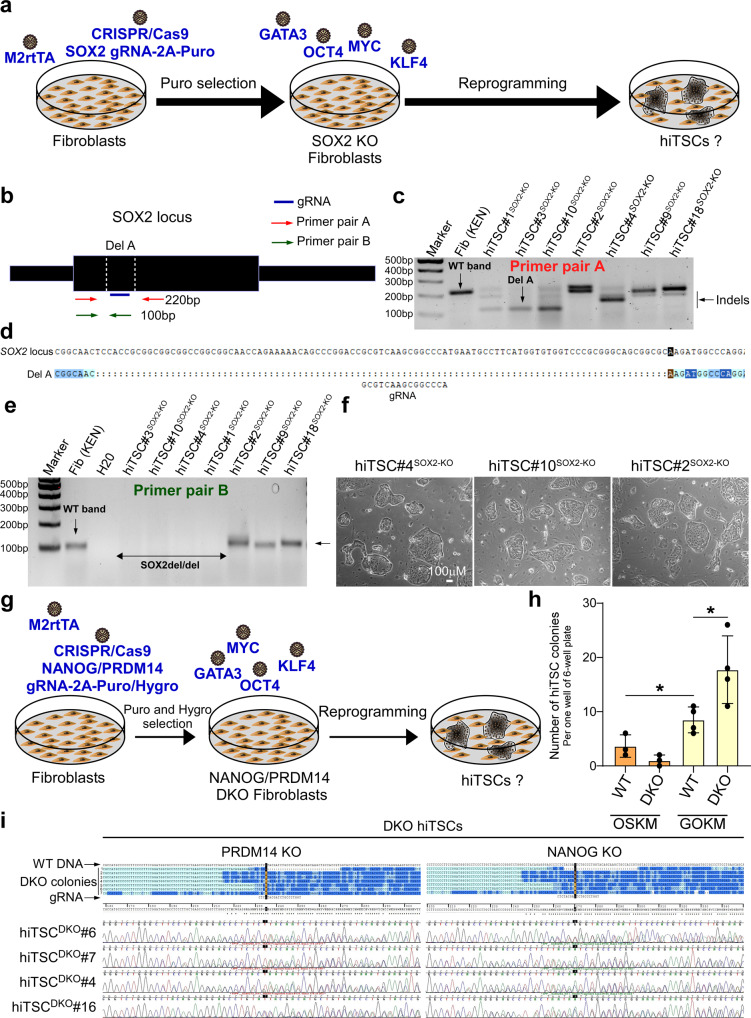
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
