

An integrated cell atlas of the lung in health and disease
- PMID: 37291214
- PMCID: PMC10287567
- DOI: 10.1038/s41591-023-02327-2
An integrated cell atlas of the lung in health and disease
Abstract
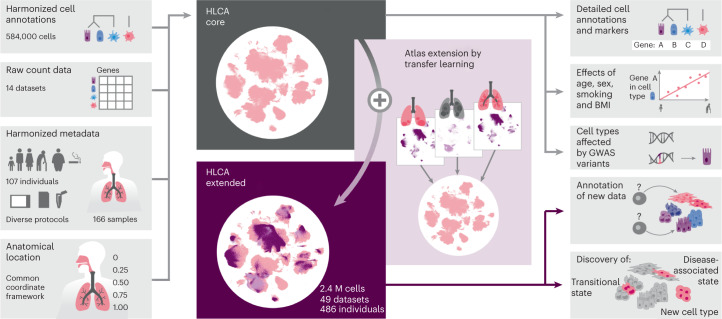
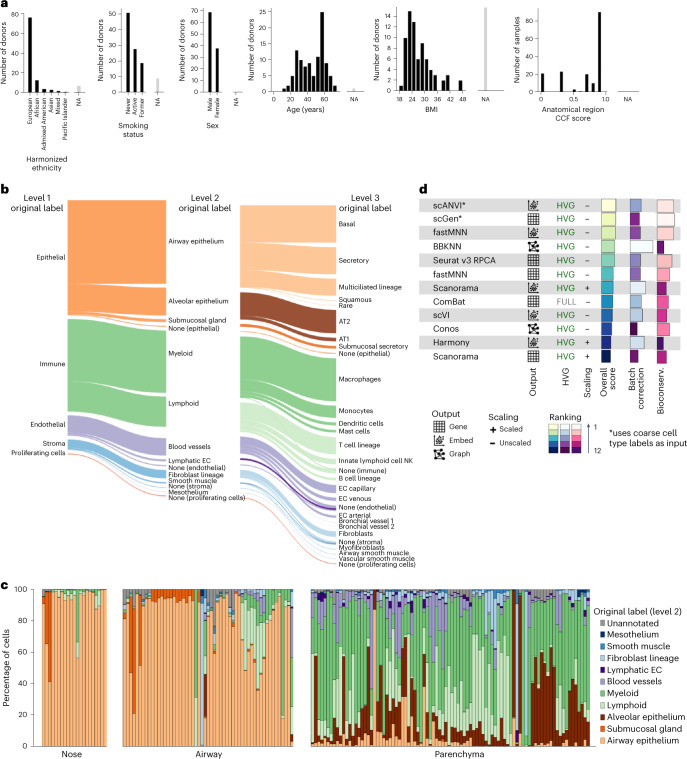
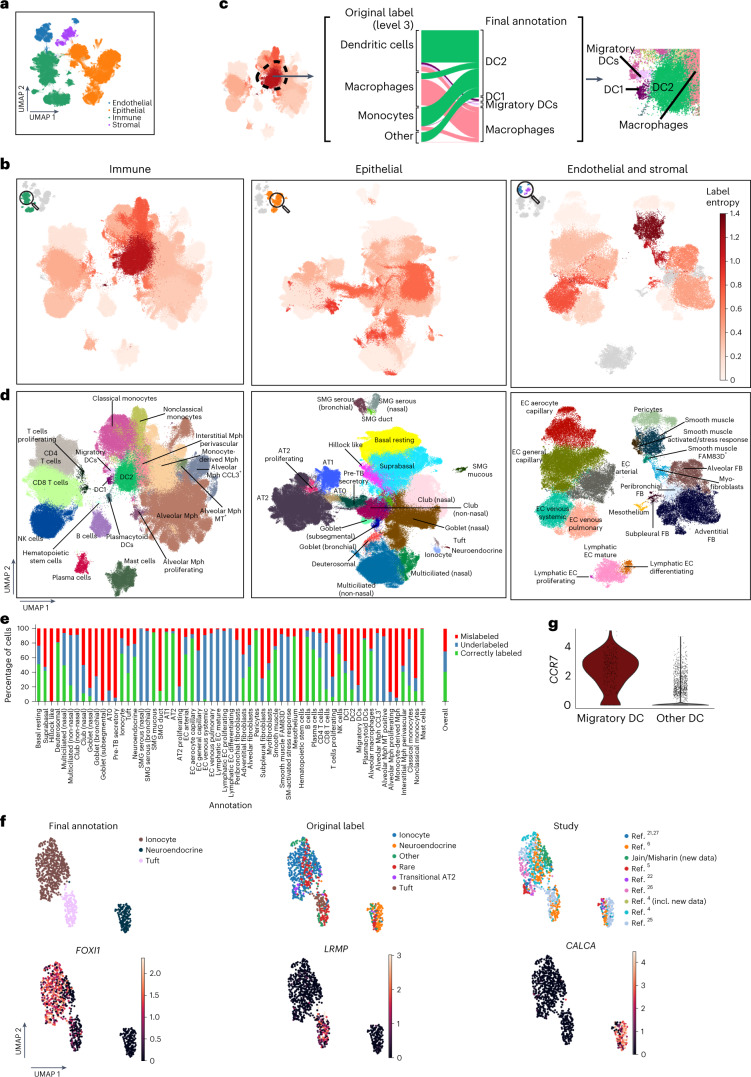
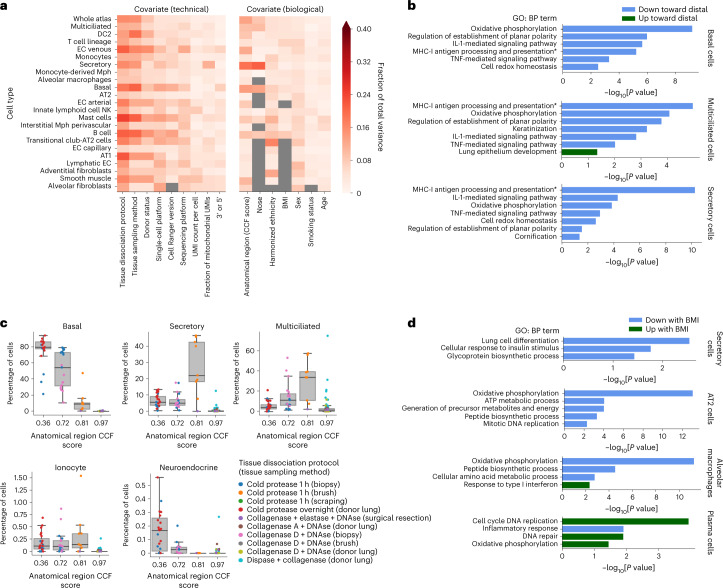
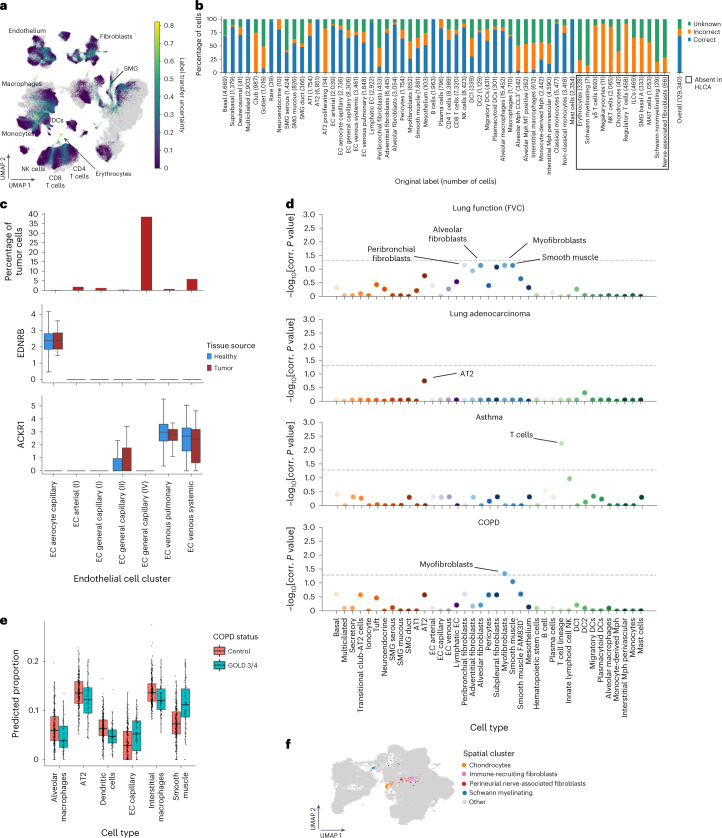
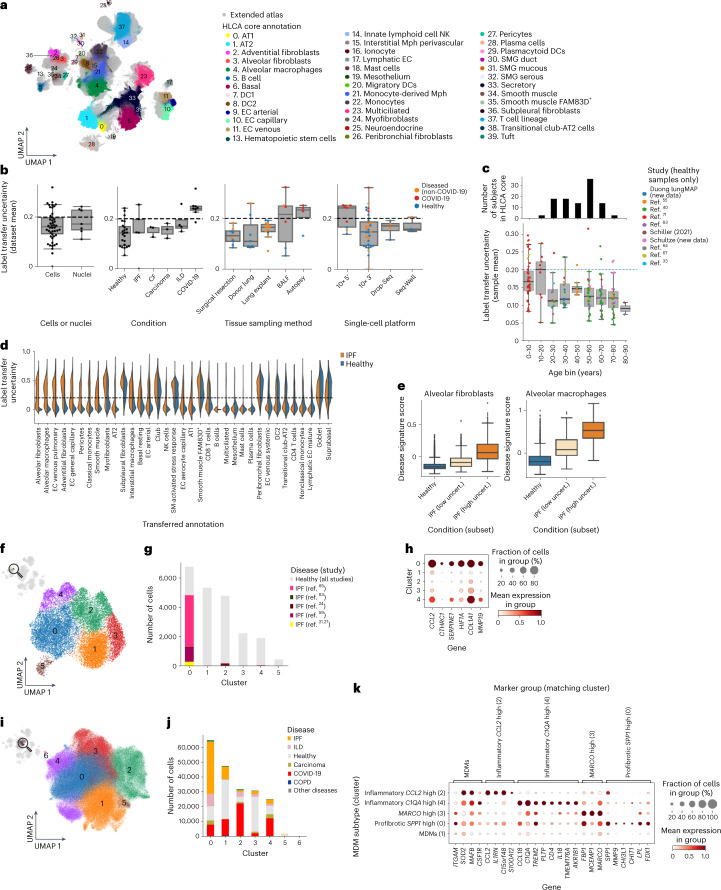
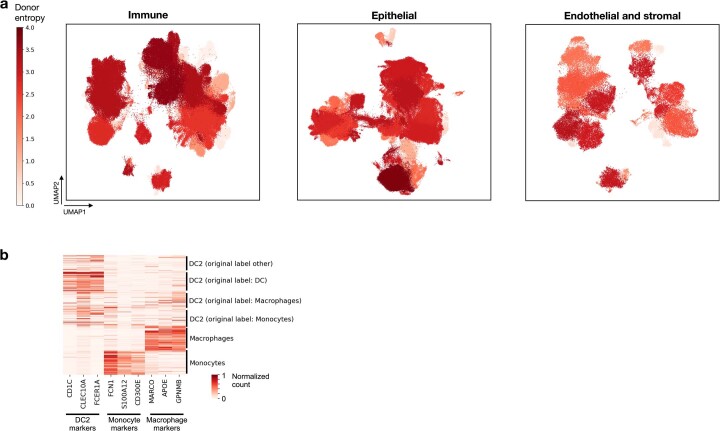
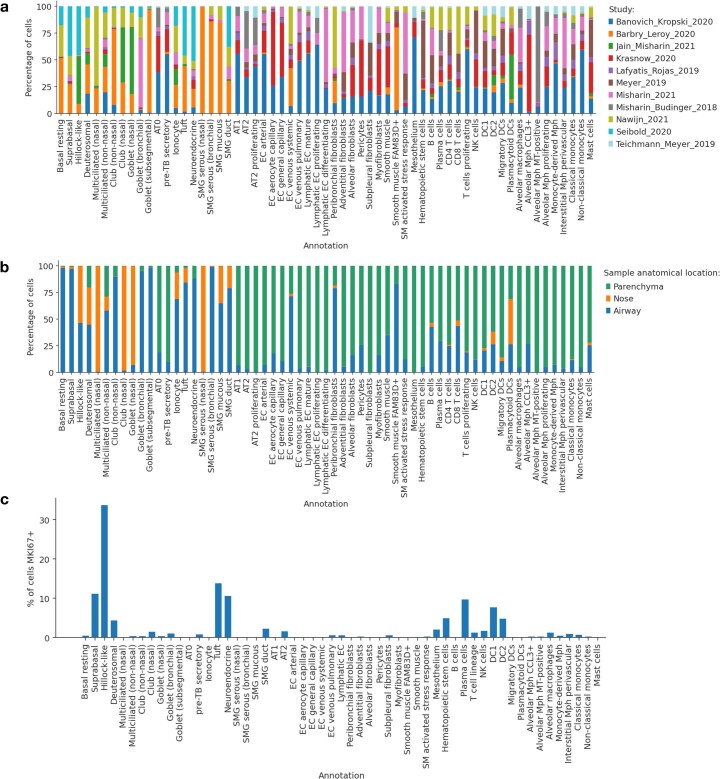
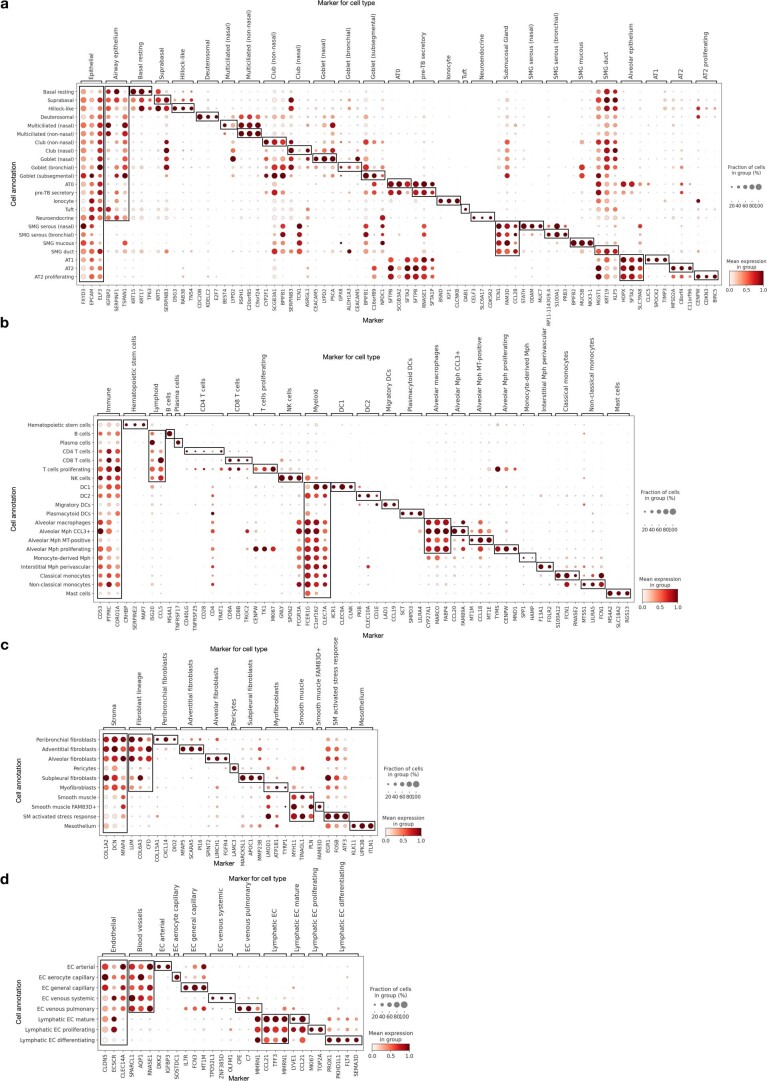
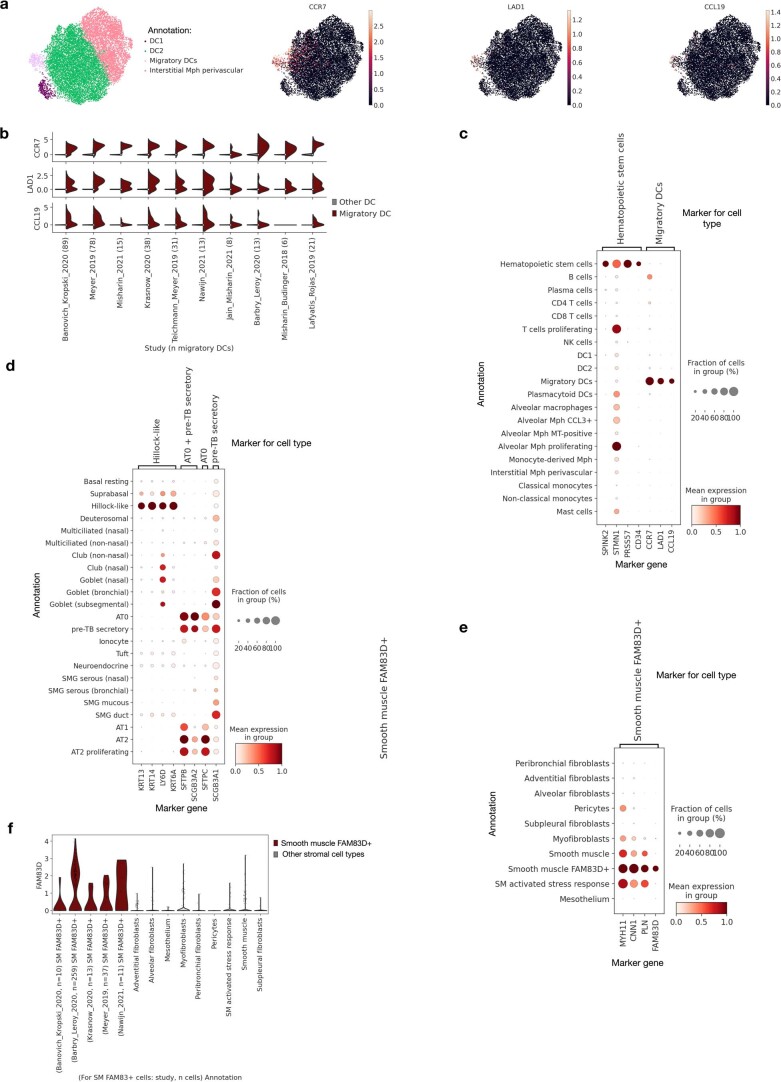
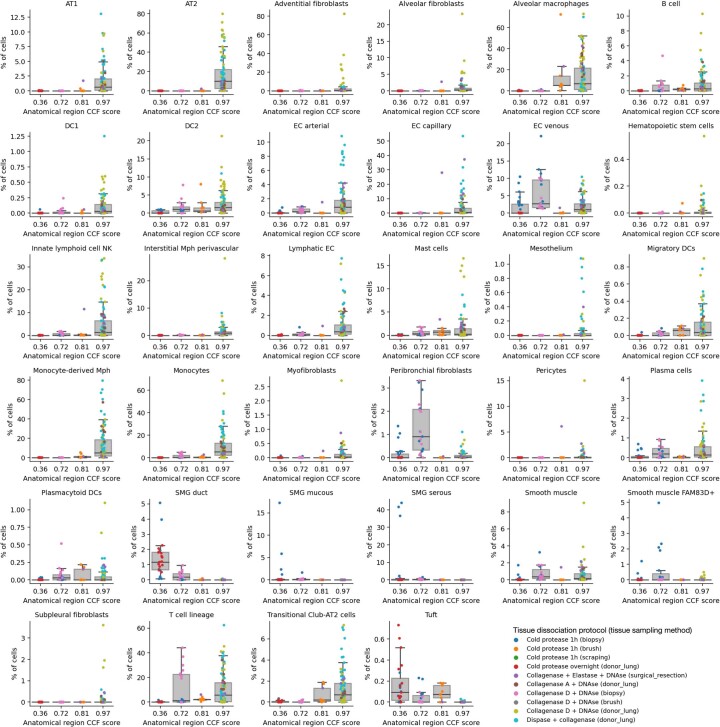
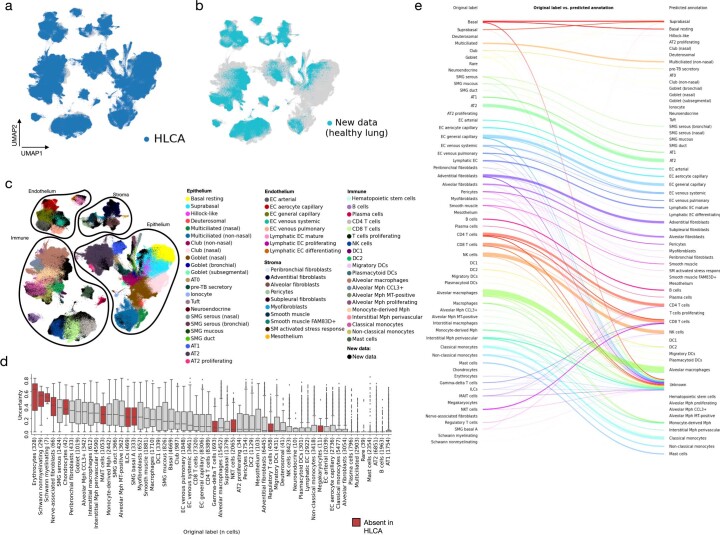
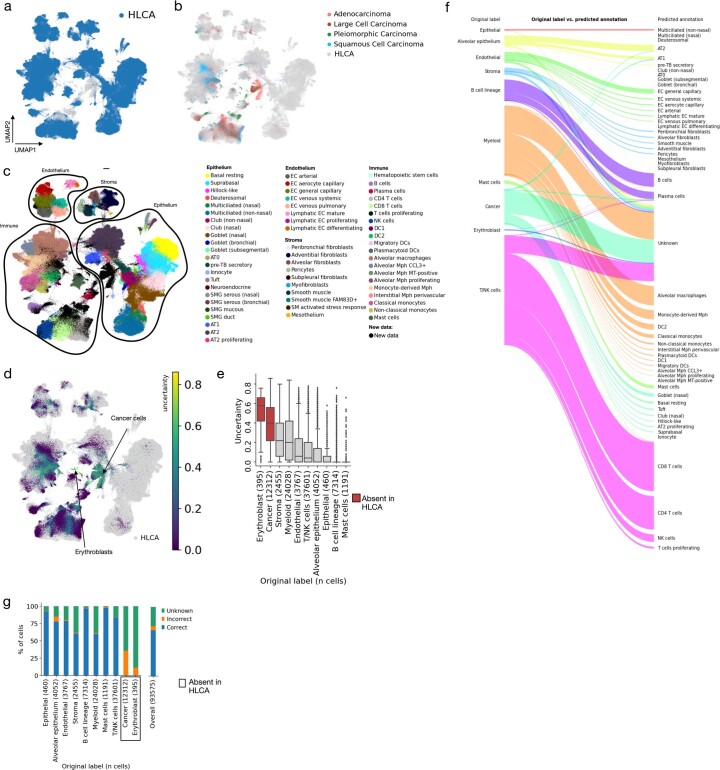
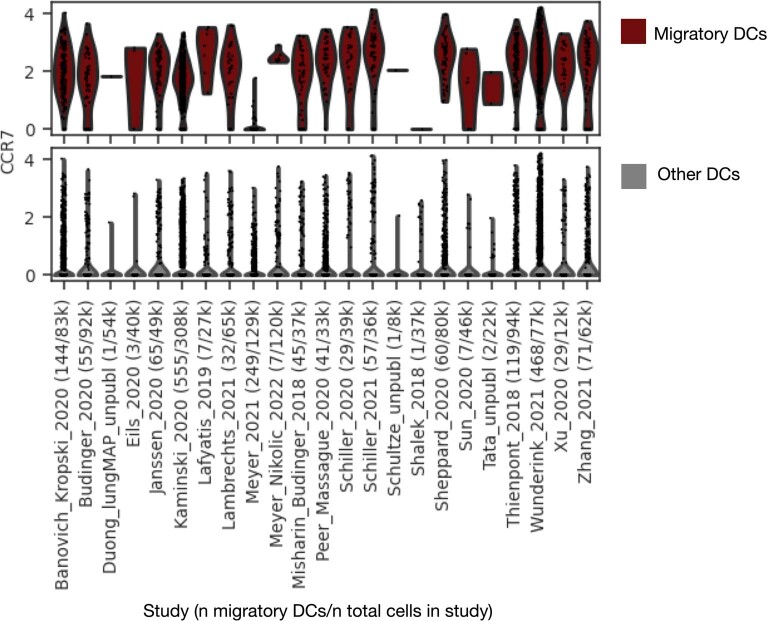
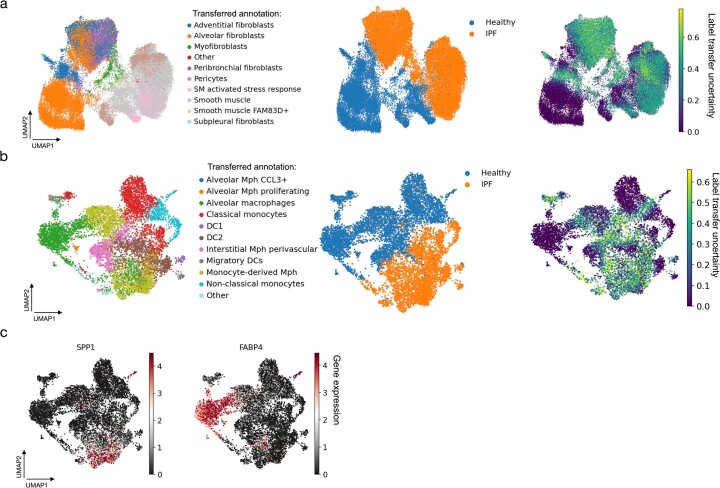
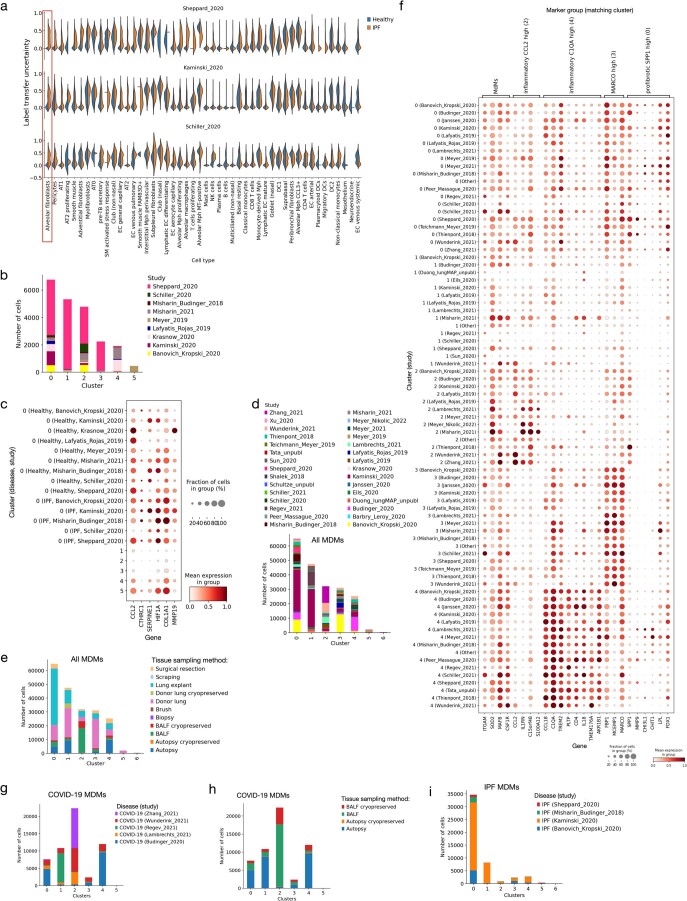
Single-cell technologies have transformed our understanding of human tissues. Yet, studies typically capture only a limited number of donors and disagree on cell type definitions. Integrating many single-cell datasets can address these limitations of individual studies and capture the variability present in the population. Here we present the integrated Human Lung Cell Atlas (HLCA), combining 49 datasets of the human respiratory system into a single atlas spanning over 2.4 million cells from 486 individuals. The HLCA presents a consensus cell type re-annotation with matching marker genes, including annotations of rare and previously undescribed cell types. Leveraging the number and diversity of individuals in the HLCA, we identify gene modules that are associated with demographic covariates such as age, sex and body mass index, as well as gene modules changing expression along the proximal-to-distal axis of the bronchial tree. Mapping new data to the HLCA enables rapid data annotation and interpretation. Using the HLCA as a reference for the study of disease, we identify shared cell states across multiple lung diseases, including SPP1+ profibrotic monocyte-derived macrophages in COVID-19, pulmonary fibrosis and lung carcinoma. Overall, the HLCA serves as an example for the development and use of large-scale, cross-dataset organ atlases within the Human Cell Atlas.
© 2023. The Author(s).
Conflict of interest statement
P.R.T. serves as a consultant for Surrozen, Cellarity and Celldom and is currently acting Chief Executive Officer of Iolux. F.J.T. consults for Immunai, Singularity Bio, CytoReason and Omniscope and has ownership interest in Dermagnostix and Cellarity. In the past 3 years, M.D.L. was a contractor for the CZI and received remuneration for talks at Pfizer and Janssen Pharmaceuticals. J.A.K. reports grants/contracts from Boehringer Ingelheim and Bristol Myers Squibb, consulting fees from Janssen and Boehringer Ingelheim and study support from Genentech and is a member of the scientific advisory board of APIE Therapeutics. In the past 3 years, S.A.T. has received remuneration for consulting and Scientific Advisory Board membership from Genentech, Roche, Biogen, GlaxoSmithKline, Foresite Labs and Qiagen. S.A.T. is a co-founder and board member of and holds equity in Transition Bio. D.S. is a founder of Pliant Therapeutics and a member of the Genentech Scientific Advisory Board and has a sponsored research agreement with AbbVie. N.K. served as a consultant to Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Theravance, LifeMax, Three Lakes Partners, Optikira, AstraZeneca, RohBar, Veracyte, Augmanity, CSL Behring, Galapagos and Thyron over the past 3 years and reports equity in Pliant and Thyron, grants from Veracyte, Boehringer Ingelheim and Bristol Myers Squibb and nonfinancial support from miRagen and AstraZeneca. N.K. owns intellectual property on novel biomarkers and therapeutics in IPF licensed to biotechnology. O.R.-R. is a co-inventor on patent applications (PCT/US2016/059233, PCT/US2018/064553, PCT/US2018/060860, PCT/US2017/016146, PCT/US2019/055894, PCT/US2018/064563, PCT/US2020/032933) filed by the Broad Institute for inventions related to single-cell genomics. O.R.-R. has been an employee of Genentech since 19 October 2020 and has equity in Roche. O.E. serves in an advisory capacity to Pieris Pharmaceuticals, Blade Therapeutics, Delta 4 and YAP Therapeutics. Y.B. holds a Canada Research Chair in the Genomics of Heart and Lung Diseases. The remaining authors declare no competing interests.
Figures
References
-
- Angerer P, et al. Single cells make big data: new challenges and opportunities in transcriptomics. Curr. Opin. Syst. Biol. 2017;4:85–91. doi: 10.1016/j.coisb.2017.07.004. - DOI
Publication types
MeSH terms
Grants and funding
- R01 HL141380/HL/NHLBI NIH HHS/United States
- P50 AR080612/AR/NIAMS NIH HHS/United States
- R01 HL153375/HL/NHLBI NIH HHS/United States
- R01 HL127349/HL/NHLBI NIH HHS/United States
- U54 HL165443/HL/NHLBI NIH HHS/United States
- P01 HL107202/HL/NHLBI NIH HHS/United States
- U01 HL148856/HL/NHLBI NIH HHS/United States
- R21 HL156124/HL/NHLBI NIH HHS/United States
- U54 AG075931/AG/NIA NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 HL146557/HL/NHLBI NIH HHS/United States
- R01 HL123766/HL/NHLBI NIH HHS/United States
- MR/R015635/1/MRC_/Medical Research Council/United Kingdom
- U01 HL148861/HL/NHLBI NIH HHS/United States
- R01 HL141852/HL/NHLBI NIH HHS/United States
- R01 ES034350/ES/NIEHS NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- R01 HL126176/HL/NHLBI NIH HHS/United States
- R21 HL161760/HL/NHLBI NIH HHS/United States
- R01 HL145372/HL/NHLBI NIH HHS/United States
- P01 AG049665/AG/NIA NIH HHS/United States
- K12 HD105271/HD/NICHD NIH HHS/United States
- U19 AI135964/AI/NIAID NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 HL142568/HL/NHLBI NIH HHS/United States
- R01 HL153312/HL/NHLBI NIH HHS/United States
- U54 AG079754/AG/NIA NIH HHS/United States
- R56 HL157632/HL/NHLBI NIH HHS/United States
- R01 HL158139/HL/NHLBI NIH HHS/United States
- R01 HL135156/HL/NHLBI NIH HHS/United States
- R01 HL153045/HL/NHLBI NIH HHS/United States
- U54 HL145608/HL/NHLBI NIH HHS/United States
- P50 AR060780/AR/NIAMS NIH HHS/United States
- R01 HL128439/HL/NHLBI NIH HHS/United States
- R01 HL146519/HL/NHLBI NIH HHS/United States
- R01 HL117004/HL/NHLBI NIH HHS/United States
- R01 HL068702/HL/NHLBI NIH HHS/United States
- U01 HL145567/HL/NHLBI NIH HHS/United States
- P01 HL132821/HL/NHLBI NIH HHS/United States
- R01 MD010443/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
