

Using machine learning to detect coronaviruses potentially infectious to humans
- PMID: 37291260
- PMCID: PMC10248971
- DOI: 10.1038/s41598-023-35861-7
Using machine learning to detect coronaviruses potentially infectious to humans
Abstract
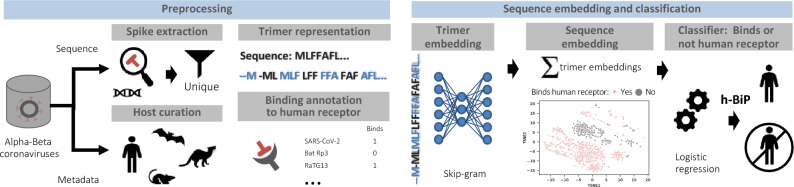
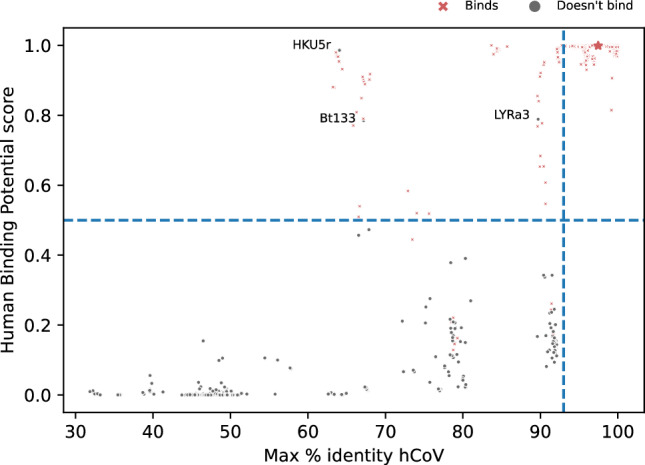
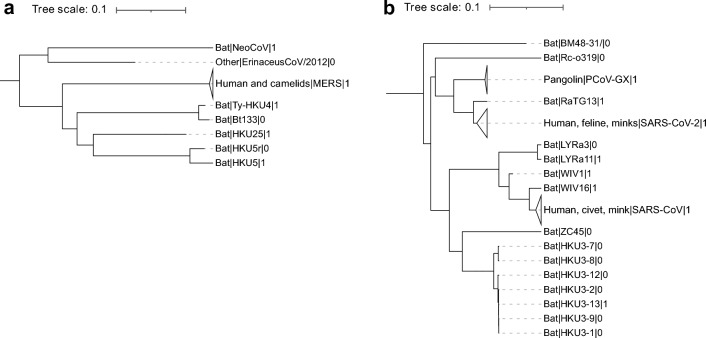
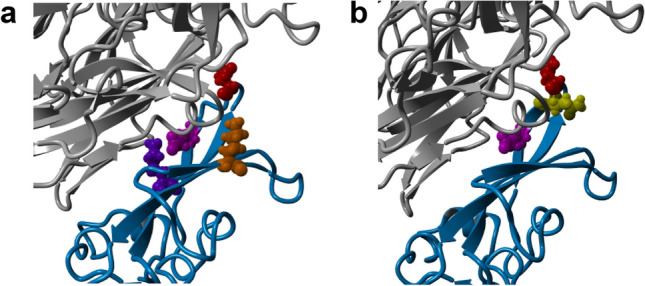
Establishing the host range for novel viruses remains a challenge. Here, we address the challenge of identifying non-human animal coronaviruses that may infect humans by creating an artificial neural network model that learns from spike protein sequences of alpha and beta coronaviruses and their binding annotation to their host receptor. The proposed method produces a human-Binding Potential (h-BiP) score that distinguishes, with high accuracy, the binding potential among coronaviruses. Three viruses, previously unknown to bind human receptors, were identified: Bat coronavirus BtCoV/133/2005 and Pipistrellus abramus bat coronavirus HKU5-related (both MERS related viruses), and Rhinolophus affinis coronavirus isolate LYRa3 (a SARS related virus). We further analyze the binding properties of BtCoV/133/2005 and LYRa3 using molecular dynamics. To test whether this model can be used for surveillance of novel coronaviruses, we re-trained the model on a set that excludes SARS-CoV-2 and all viral sequences released after the SARS-CoV-2 was published. The results predict the binding of SARS-CoV-2 with a human receptor, indicating that machine learning methods are an excellent tool for the prediction of host expansion events.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Rodriguez-Morales AJ, et al. History is repeating itself: Probable zoonotic spillover as the cause of the 2019 novel Coronavirus Epidemic. Infez. Med. 2020;28(1):3–5. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
