

Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR
- PMID: 37291434
- PMCID: PMC10504084
- DOI: 10.1038/s41551-023-01048-8
Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR
Abstract
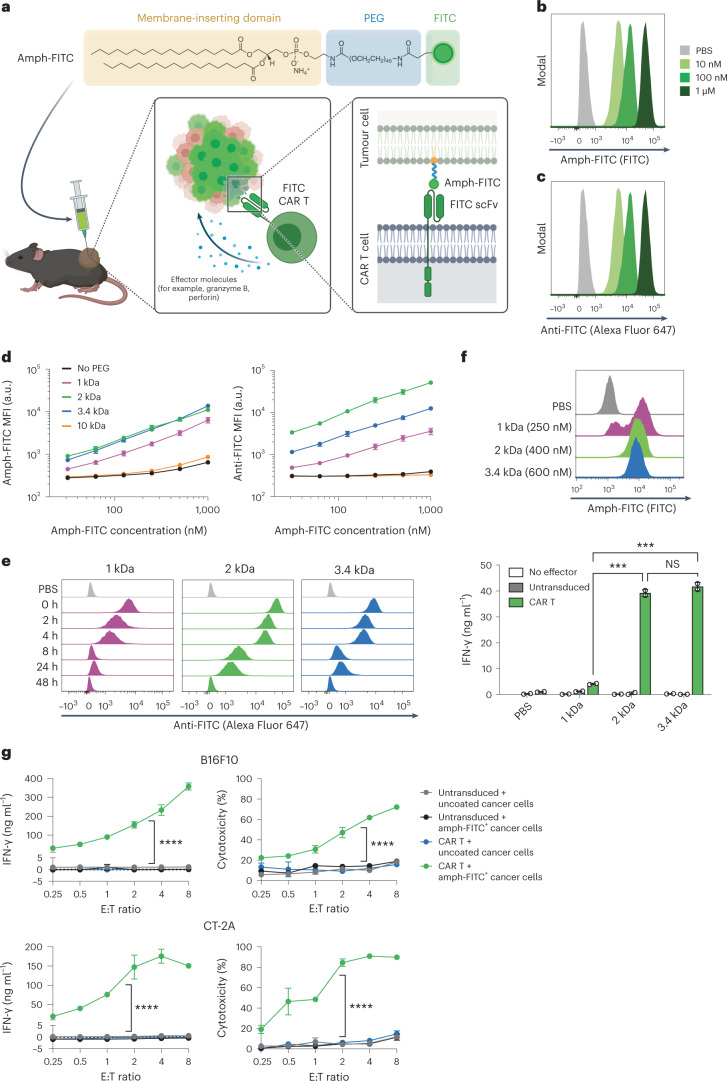
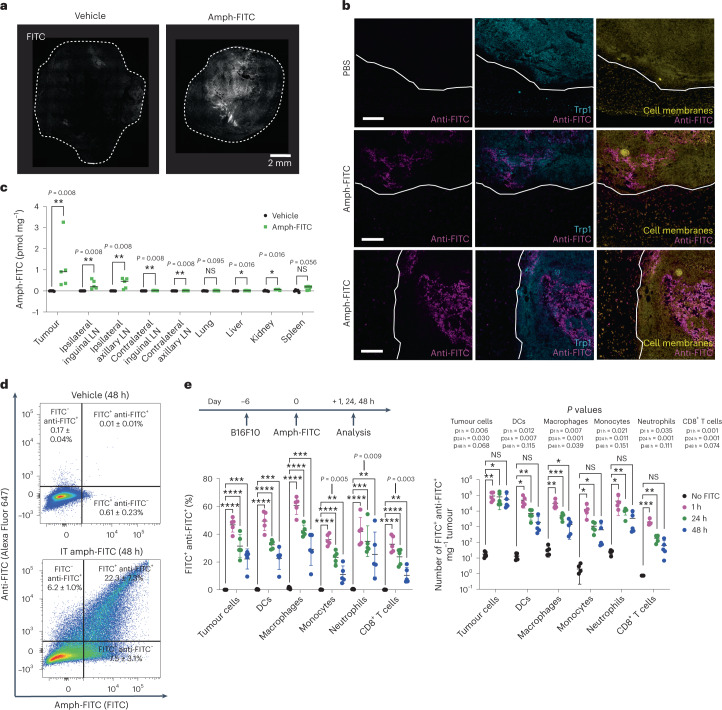
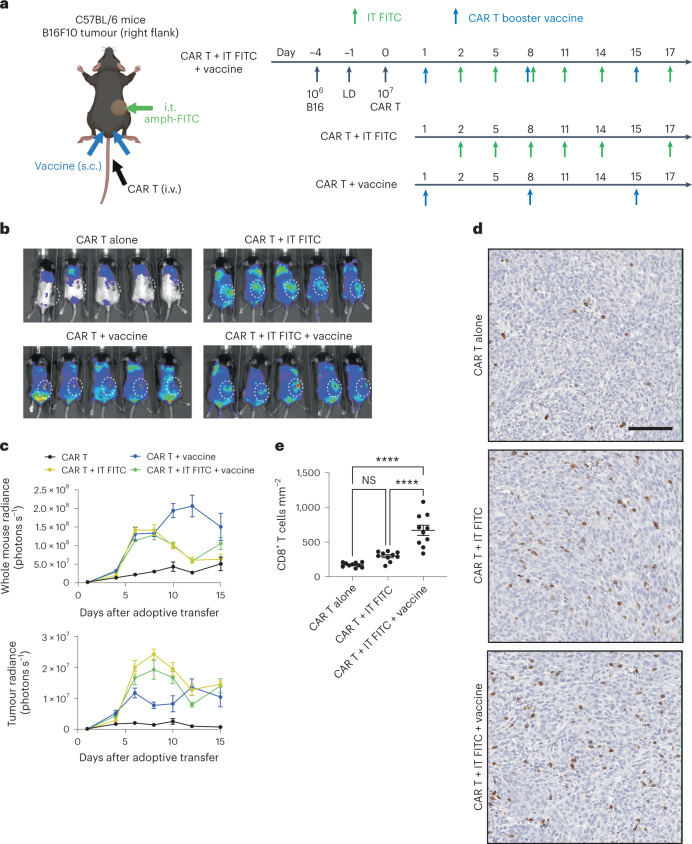
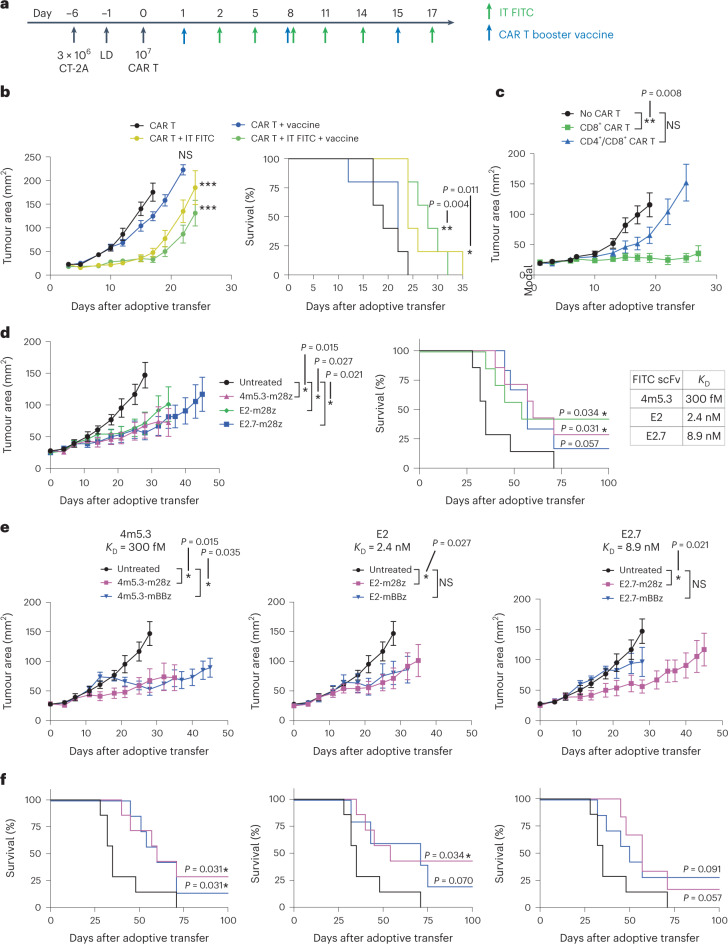
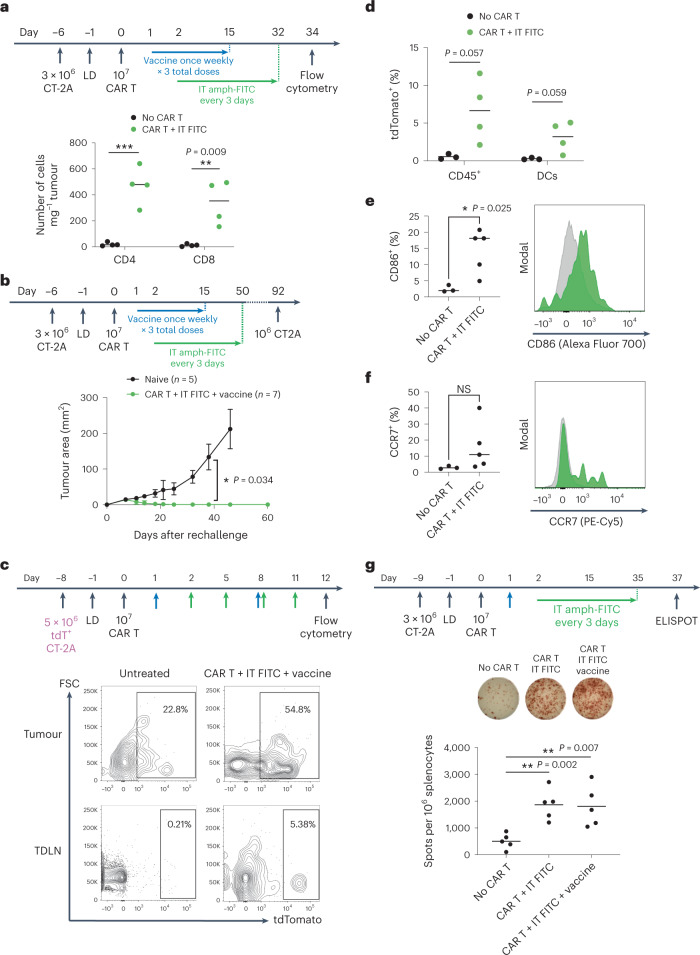
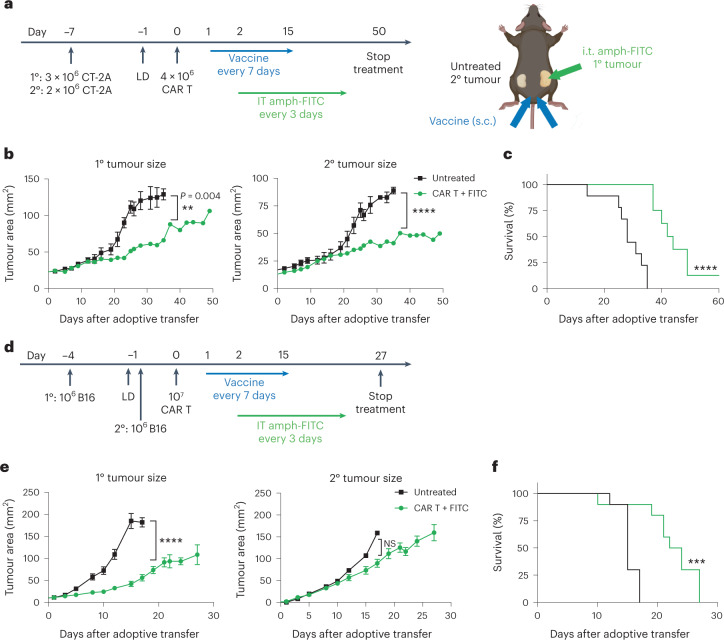
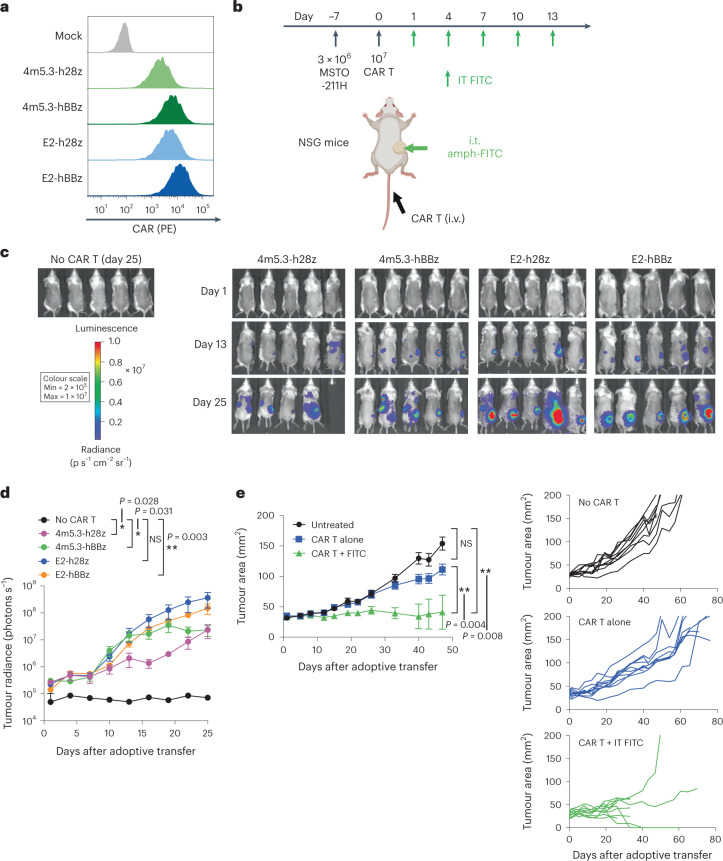
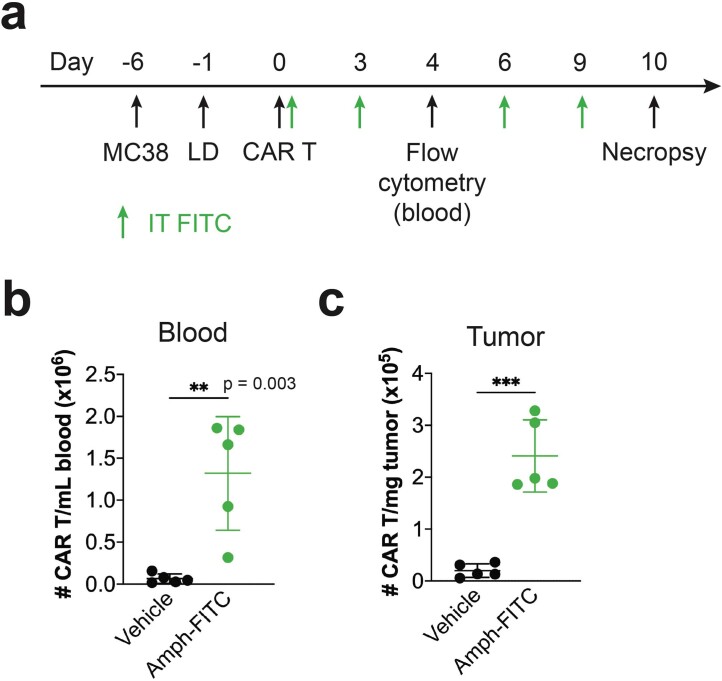
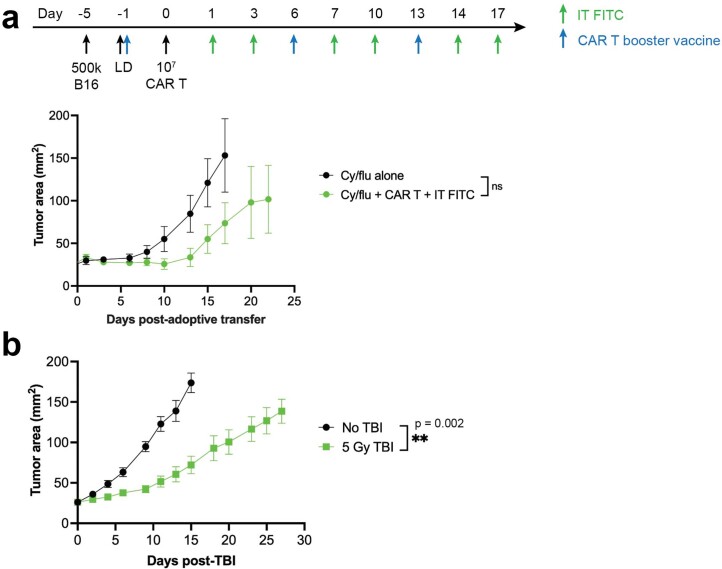
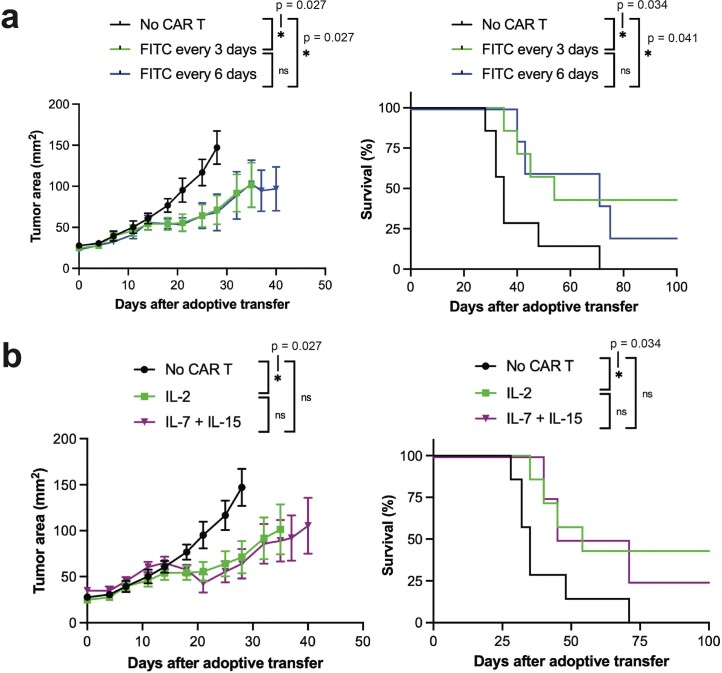
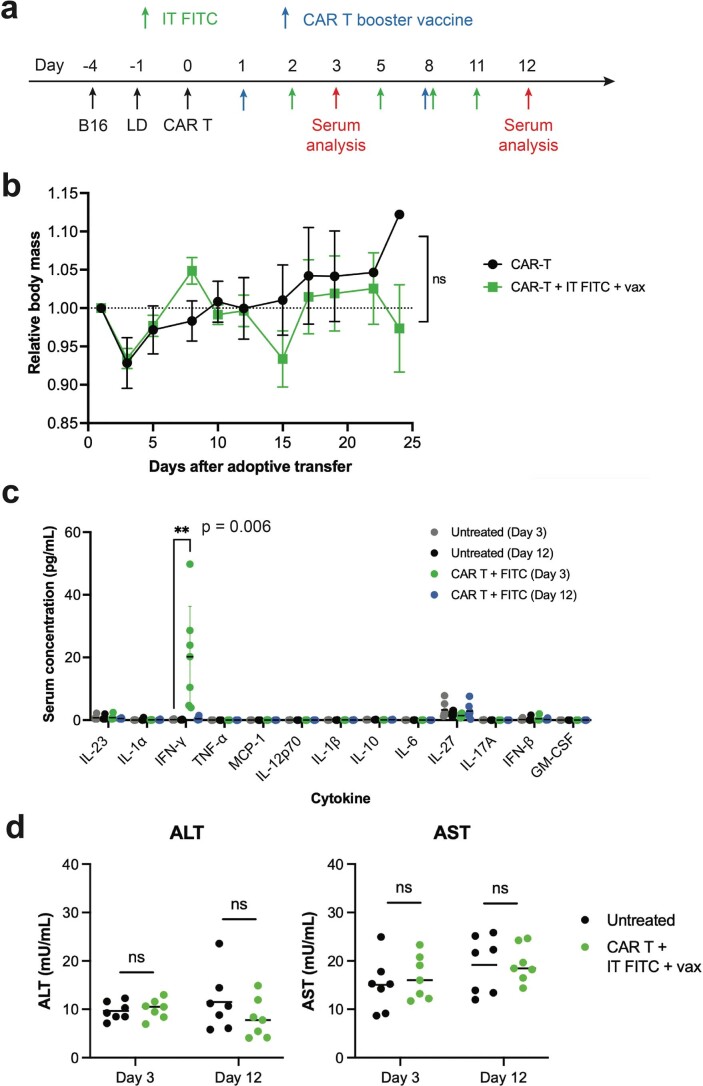
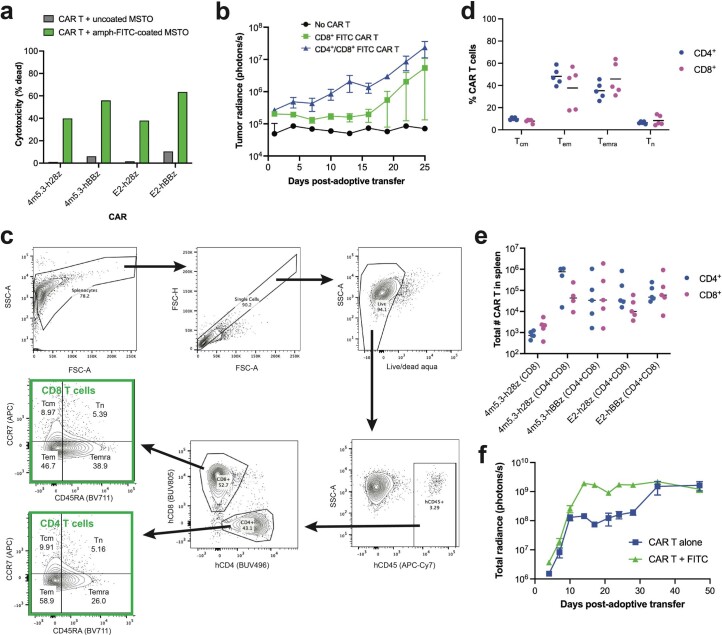
The effectiveness of chimaeric antigen receptor (CAR) T cell therapies for solid tumours is hindered by difficulties in the selection of an effective target antigen, owing to the heterogeneous expression of tumour antigens and to target antigen expression in healthy tissues. Here we show that T cells with a CAR specific for fluorescein isothiocyanate (FITC) can be directed against solid tumours via the intratumoural administration of a FITC-conjugated lipid-poly(ethylene)-glycol amphiphile that inserts itself into cell membranes. In syngeneic and human tumour xenografts in mice, 'amphiphile tagging' of tumour cells drove tumour regression via the proliferation and accumulation of FITC-specific CAR T cells in the tumours. In syngeneic tumours, the therapy induced the infiltration of host T cells, elicited endogenous tumour-specific T cell priming and led to activity against distal untreated tumours and to protection against tumour rechallenge. Membrane-inserting ligands for specific CARs may facilitate the development of adoptive cell therapies that work independently of antigen expression and of tissue of origin.
© 2023. The Author(s).
Conflict of interest statement
A.Q.Z., A.H., L.E.C. and D.J.I. have submitted a patent application filed by MIT related to the data presented in this work. D.J.I. is a consultant and equity holder in Elicio Therapeutics, which has licensed rights to the MIT intellectual property mentioned above. The other authors declare no interests.
Figures
References
-
- Mullard A. FDA approves fourth CAR-T cell therapy. Nature. 2021;20:166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
