

This is a preprint.
CRISPR-based engineering of RNA viruses
- PMID: 37292641
- PMCID: PMC10245796
- DOI: 10.1101/2023.05.19.541219
CRISPR-based engineering of RNA viruses
Update in
-
CRISPR-based engineering of RNA viruses.Sci Adv. 2023 Sep 15;9(37):eadj8277. doi: 10.1126/sciadv.adj8277. Epub 2023 Sep 13. Sci Adv. 2023. PMID: 37703376 Free PMC article.
Abstract
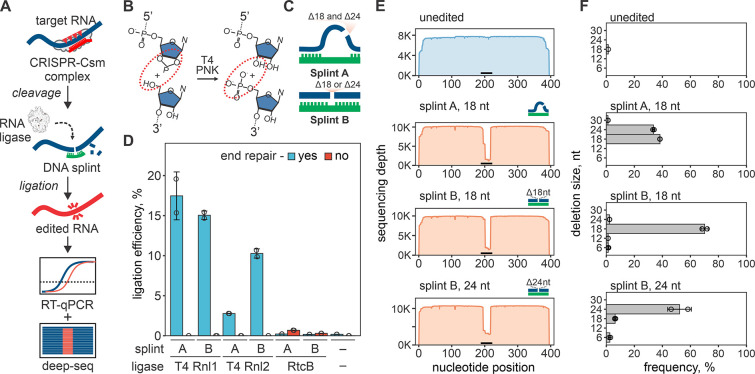
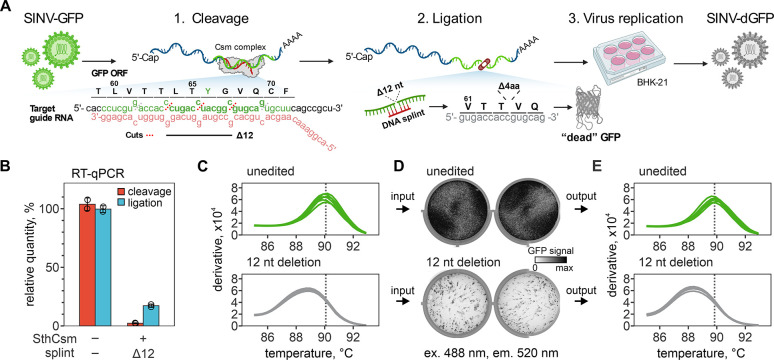
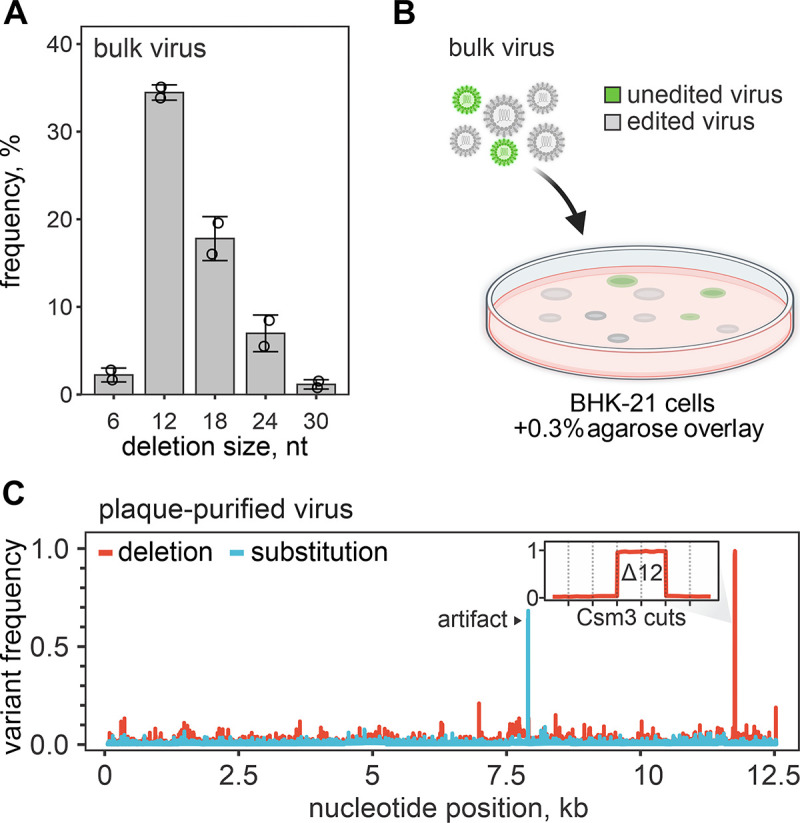
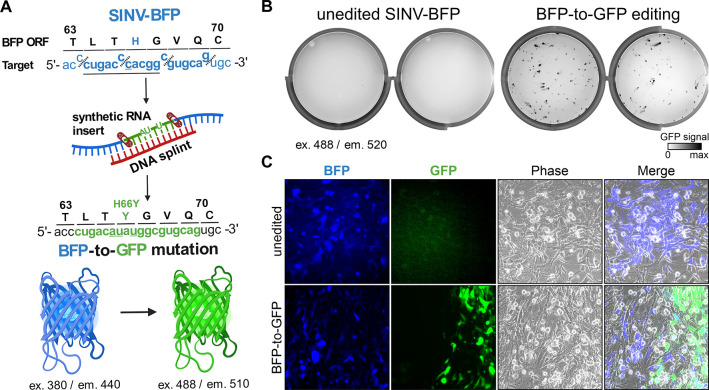
CRISPR RNA-guided endonucleases have enabled precise editing of DNA. However, options for editing RNA remain limited. Here, we combine sequence-specific RNA cleavage by CRISPR ribonucleases with programmable RNA repair to make precise deletions and insertions in RNA. This work establishes a new recombinant RNA technology with immediate applications for the facile engineering of RNA viruses.
One-sentence summary: Programmable CRISPR RNA-guided ribonucleases enable recombinant RNA technology.
Conflict of interest statement
Figures
References
-
- Jackson D. A., Symons R. H., Berg P., Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proceedings of the National Academy of Sciences 69, 2904–2909 (1972). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources