

This is a preprint.
Suppression of tumor cell lactate-generating signaling pathways eradicates murine PTEN/p53-deficient aggressive-variant prostate cancer via macrophage phagocytosis
- PMID: 37292972
- PMCID: PMC10245812
- DOI: 10.1101/2023.05.23.540590
Suppression of tumor cell lactate-generating signaling pathways eradicates murine PTEN/p53-deficient aggressive-variant prostate cancer via macrophage phagocytosis
Update in
-
Suppression of Tumor Cell Lactate-generating Signaling Pathways Eradicates Murine PTEN/p53-deficient Aggressive-variant Prostate Cancer via Macrophage Phagocytosis.Clin Cancer Res. 2023 Dec 1;29(23):4930-4940. doi: 10.1158/1078-0432.CCR-23-1441. Clin Cancer Res. 2023. PMID: 37721526 Free PMC article.
Abstract
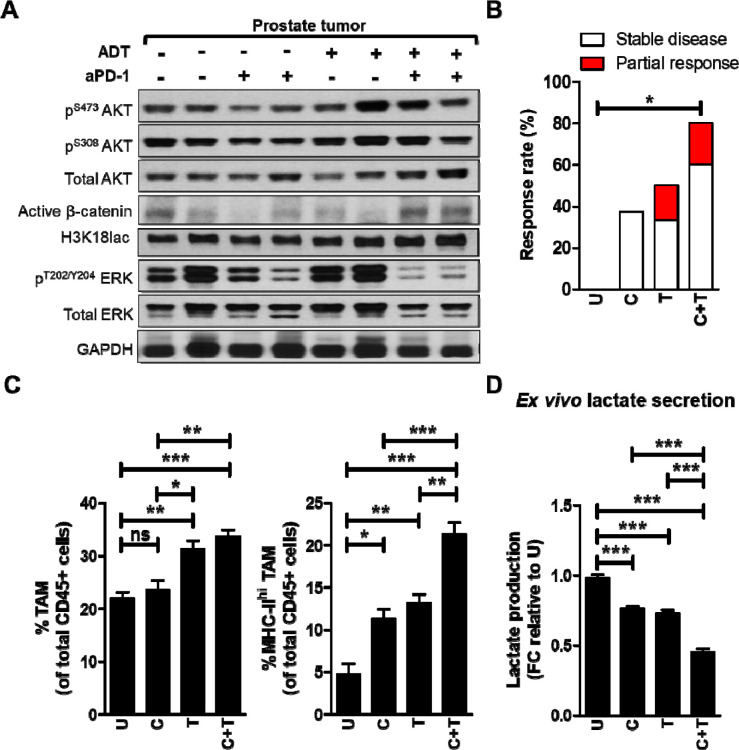
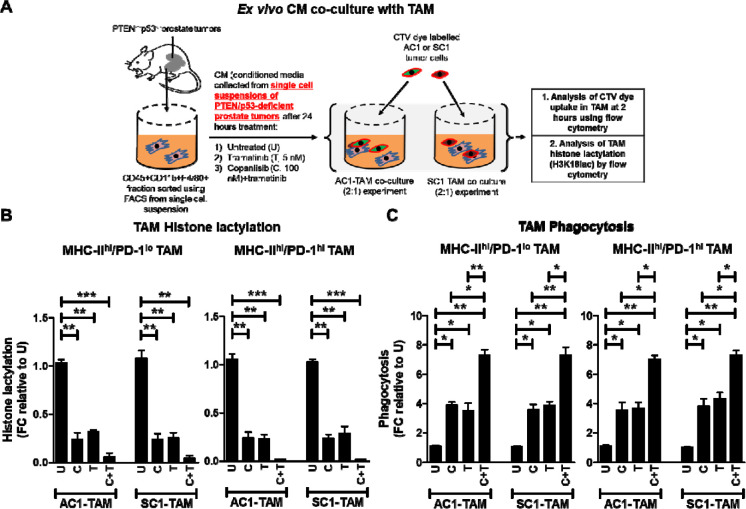
Purpose: PTEN loss-of-function/PI3K pathway hyperactivation occurs in ∼50% of metastatic, castrate-resistant prostate cancer patients, resulting in poor therapeutic outcomes and resistance to immune checkpoint inhibitors across multiple malignancies. Our prior studies in prostate-specific PTEN/p53-deleted genetically engineered mice (Pb-Cre;PTEN fl/fl Trp53 fl/fl GEM) with aggressive-variant prostate cancer (AVPC) demonstrated feedback Wnt/β-catenin signaling activation in 40% mice resistant to androgen deprivation therapy (ADT)/PI3K inhibitor (PI3Ki)/PD-1 antibody (aPD-1) combination, resulting in restoration of lactate cross-talk between tumor-cells and tumor-associated macrophages (TAM), histone lactylation (H3K18lac) and phagocytic suppression within TAM. Here, we targeted immunometabolic mechanism(s) of resistance to ADT/PI3Ki/aPD-1 combination, with the goal of durable tumor control in PTEN/p53-deficient PC.
Experimental design: Pb-Cre;PTEN fl/fl Trp53 fl/fl GEM were treated with either ADT (degarelix), PI3Ki (copanlisib), aPD-1, MEK inhibitor (trametinib) or Porcupine inhibitor (LGK 974) as single agents or their combinations. MRI was used to monitor tumor kinetics and immune/proteomic profiling/ ex vivo co-culture mechanistic studies were performed on prostate tumors or established GEM-derived cell lines.
Results: We tested whether Wnt/β-catenin pathway inhibition with LGK 974 addition to degarelix/copanlisib/aPD-1 therapy enhances tumor control in GEM, and observed de novo resistance due to feedback activation of MEK signaling. Based on our observation that degarelix/aPD-1 treatment resulted in partial inhibition of MEK signaling, we substituted trametinib for degarelix/aPD-1 treatment, and observed a durable tumor growth control of PI3Ki/MEKi/PORCNi in 100% mice via H3K18lac suppression and complete TAM activation within TME.
Conclusions: Abrogation of lactate-mediated cross-talk between cancer cells and TAM results in durable ADT-independent tumor control in PTEN/p53-deficient AVPC, and warrants further investigation in clinical trials.
Statement of translational relevance: PTEN loss-of-function occurs in ∼50% of mCRPC patients, and associated with poor prognosis, and immune checkpoint inhibitor resistance across multiple malignancies. Our prior studies have demonstrated that ADT/PI3Ki/PD-1 triplet combination therapy controls PTEN/p53-deficient PC in 60% of mice via enhancement of TAM phagocytosis. Here, we discovered that resistance to ADT/PI3K/PD-1 therapy occurred via restoration of lactate production via feedback Wnt/MEK signaling following treatment with PI3Ki, resulting in inhibition of TAM phagocytosis. Critically, co-targeting of PI3K/MEK/Wnt signaling pathways using an intermittent dosing schedule of corresponding targeted agents resulted in complete tumor control and significantly prolonged survival without significant long-term toxicity. Collectively, our findings provide "proof-of-concept" that targeting lactate as a macrophage phagocytic checkpoint controls growth of murine PTEN/p53-deficient PC and warrant further investigation in AVPC clinical trials.
Conflict of interest statement
Figures




References
-
- Li Y, Malapati S, Lin YT, Patnaik A. An integrative approach for sequencing therapies in metastatic prostate cancer. The American Journal of Hematology/Oncology 2017;13(12):26–31.
-
- Sydes MR, Spears MR, Mason MD, Clarke NW, Dearnaley DP, de Bono JS, et al. Adding abiraterone or docetaxel to long-term hormone therapy for prostate cancer: directly randomised data from the STAMPEDE multi-arm, multi-stage platform protocol. Ann Oncol 2018;29(5):1235–48 doi 10.1093/annonc/mdy072. - DOI - PMC - PubMed
-
- Clarke NW, Ali A, Ingleby FC, Hoyle A, Amos CL, Attard G, et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: long-term survival results from the STAMPEDE trial. Ann Oncol 2019;30(12):1992–2003 doi 10.1093/annonc/mdz396. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous