

This is a preprint.
Rapid and sensitive detection of genome contamination at scale with FCS-GX
- PMID: 37292984
- PMCID: PMC10246020
- DOI: 10.1101/2023.06.02.543519
Rapid and sensitive detection of genome contamination at scale with FCS-GX
Update in
-
Rapid and sensitive detection of genome contamination at scale with FCS-GX.Genome Biol. 2024 Feb 26;25(1):60. doi: 10.1186/s13059-024-03198-7. Genome Biol. 2024. PMID: 38409096 Free PMC article.
Abstract
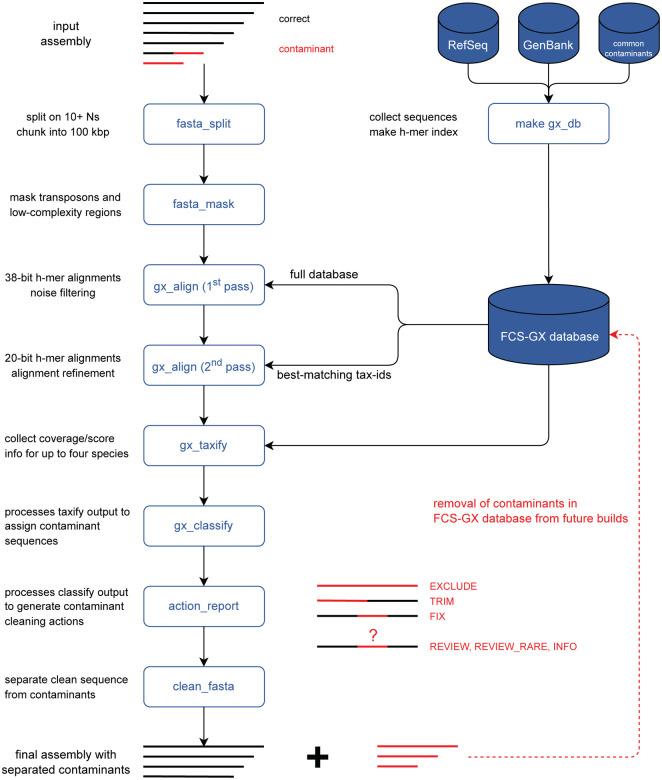
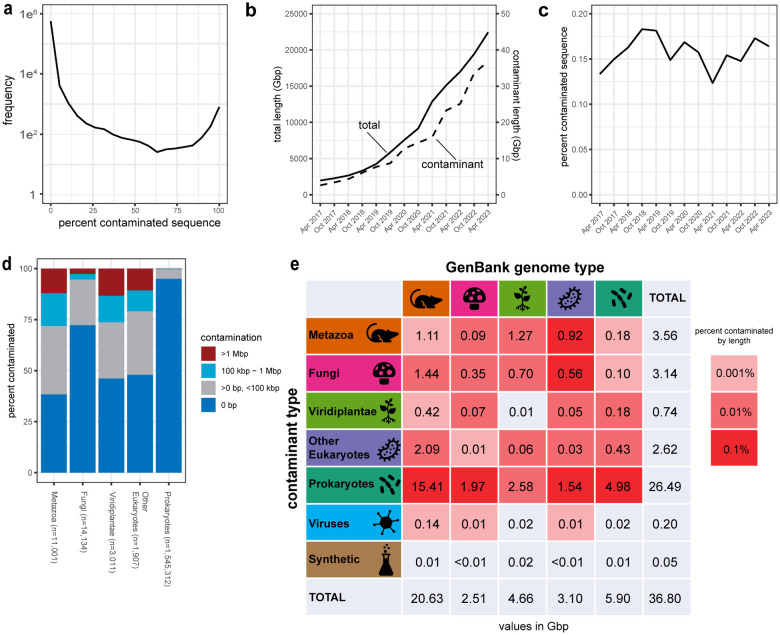
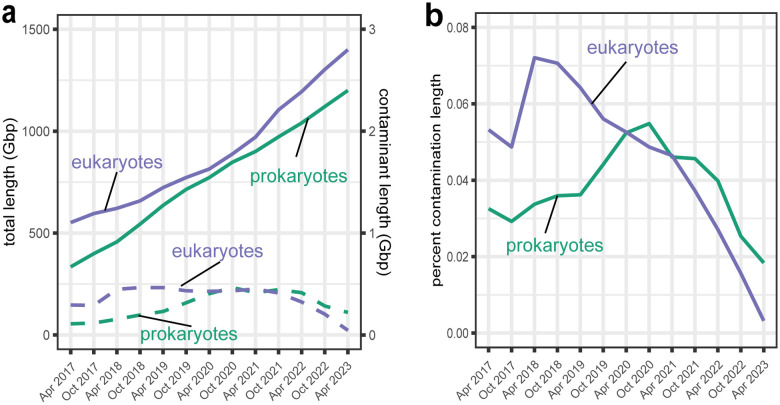
Assembled genome sequences are being generated at an exponential rate. Here we present FCS-GX, part of NCBI's Foreign Contamination Screen (FCS) tool suite, optimized to identify and remove contaminant sequences in new genomes. FCS-GX screens most genomes in 0.1-10 minutes. Testing FCS-GX on artificially fragmented genomes demonstrates sensitivity >95% for diverse contaminant species and specificity >99.93%. We used FCS-GX to screen 1.6 million GenBank assemblies and identified 36.8 Gbp of contamination (0.16% of total bases), with half from 161 assemblies. We updated assemblies in NCBI RefSeq to reduce detected contamination to 0.01% of bases. FCS-GX is available at https://github.com/ncbi/fcs/.
Keywords: GenBank; Genome assembly; Genome contamination; Genome quality; RefSeq; Software.
Conflict of interest statement
Competing interests The authors declare that they have no competing interests.
Figures

References
-
- van der Valk T, Vezzi F, Ormestad M, Dalén L, Guschanski K. Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol Ecol Resour. 2020; 20:1171–1181. - PubMed
-
- Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, Wei E, Chan CK, Nabhan AN, Su T, Morganti RM. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. BioRxiv. 2017; 10.1101/125724. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials