

This is a preprint.
Assessing Fairness of AlphaFold2 Prediction of Protein 3D Structures
- PMID: 37293014
- PMCID: PMC10245900
- DOI: 10.1101/2023.05.23.542006
Assessing Fairness of AlphaFold2 Prediction of Protein 3D Structures
Abstract
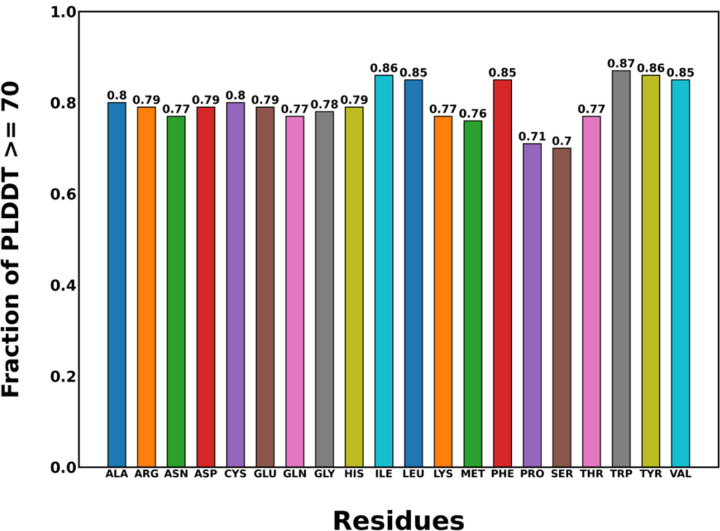
AlphaFold2 is reshaping biomedical research by enabling the prediction of a protein's 3D structure solely based on its amino acid sequence. This breakthrough reduces reliance on labor-intensive experimental methods traditionally used to obtain protein structures, thereby accelerating the pace of scientific discovery. Despite the bright future, it remains unclear whether AlphaFold2 can uniformly predict the wide spectrum of proteins equally well. Systematic investigation into the fairness and unbiased nature of its predictions is still an area yet to be thoroughly explored. In this paper, we conducted an in-depth analysis of AlphaFold2's fairness using data comprised of five million reported protein structures from its open-access repository. Specifically, we assessed the variability in the distribution of PLDDT scores, considering factors such as amino acid type, secondary structure, and sequence length. Our findings reveal a systematic discrepancy in AlphaFold2's predictive reliability, varying across different types of amino acids and secondary structures. Furthermore, we observed that the size of the protein exerts a notable impact on the credibility of the 3D structural prediction. AlphaFold2 demonstrates enhanced prediction power for proteins of medium size compared to those that are either smaller or larger. These systematic biases could potentially stem from inherent biases present in its training data and model architecture. These factors need to be taken into account when expanding the applicability of AlphaFold2.
Keywords: AI fairness; AlphaFold; protein structures.
Figures















References
-
- Anfinsen C.B., Principles that govern the folding of protein chains. Science, 1973. 181. - PubMed
-
- Pinheiro F., Santos J., and Ventura S., AlphaFold and the amyloid landscape. J Mol Biol, 2021. 433(20): p. 167059. - PubMed
-
- Fleishman S.J. and Horovitz A., Extending the New Generation of Structure Predictors to Account for Dynamics and Allostery. J Mol Biol, 2021. 433(20): p. 167007. - PubMed
Publication types
LinkOut - more resources
Full Text Sources