

Mitochondrial dysfunctions induce PANoptosis and ferroptosis in cerebral ischemia/reperfusion injury: from pathology to therapeutic potential
- PMID: 37293623
- PMCID: PMC10244524
- DOI: 10.3389/fncel.2023.1191629
Mitochondrial dysfunctions induce PANoptosis and ferroptosis in cerebral ischemia/reperfusion injury: from pathology to therapeutic potential
Abstract
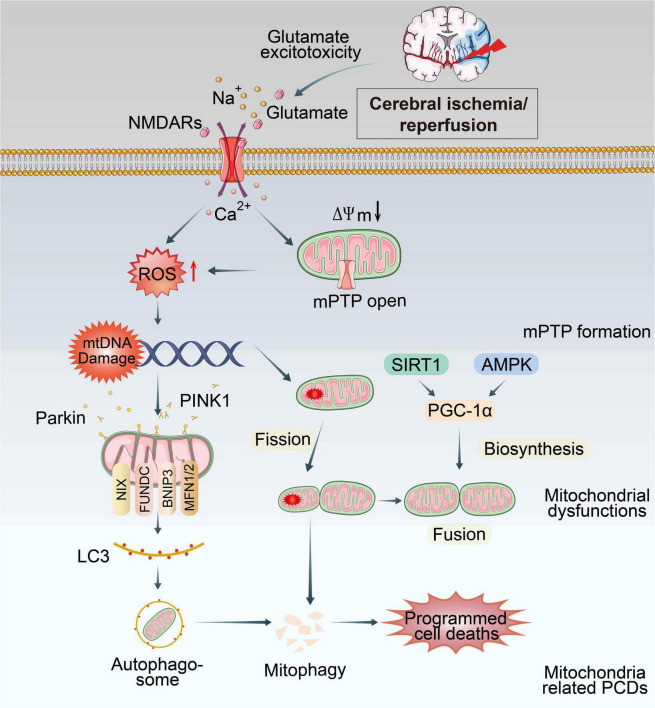
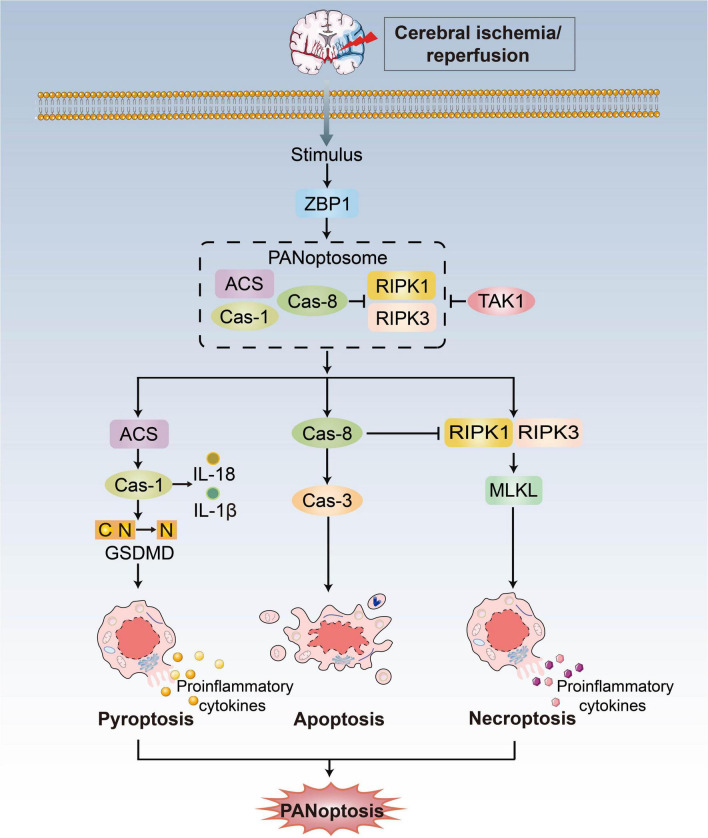
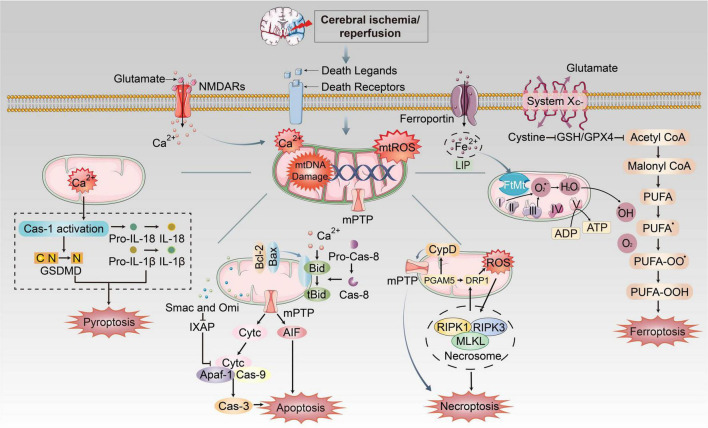
Ischemic stroke (IS) accounts for more than 80% of the total stroke, which represents the leading cause of mortality and disability worldwide. Cerebral ischemia/reperfusion injury (CI/RI) is a cascade of pathophysiological events following the restoration of blood flow and reoxygenation, which not only directly damages brain tissue, but also enhances a series of pathological signaling cascades, contributing to inflammation, further aggravate the damage of brain tissue. Paradoxically, there are still no effective methods to prevent CI/RI, since the detailed underlying mechanisms remain vague. Mitochondrial dysfunctions, which are characterized by mitochondrial oxidative stress, Ca2+ overload, iron dyshomeostasis, mitochondrial DNA (mtDNA) defects and mitochondrial quality control (MQC) disruption, are closely relevant to the pathological process of CI/RI. There is increasing evidence that mitochondrial dysfunctions play vital roles in the regulation of programmed cell deaths (PCDs) such as ferroptosis and PANoptosis, a newly proposed conception of cell deaths characterized by a unique form of innate immune inflammatory cell death that regulated by multifaceted PANoptosome complexes. In the present review, we highlight the mechanisms underlying mitochondrial dysfunctions and how this key event contributes to inflammatory response as well as cell death modes during CI/RI. Neuroprotective agents targeting mitochondrial dysfunctions may serve as a promising treatment strategy to alleviate serious secondary brain injuries. A comprehensive insight into mitochondrial dysfunctions-mediated PCDs can help provide more effective strategies to guide therapies of CI/RI in IS.
Keywords: PANoptosis; PANoptosome; cerebral ischemia/reperfusion injury (CI/RI); ferroptosis; ischemic stroke; mitochondrial dysfunctions.
Copyright © 2023 She, Liu, Liao, Wang, Ge and Mei.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous