

Terminal microdeletion of chromosome 18 in a Malaysian boy characterized with few features of typical 18q- deletion syndrome: a case report
- PMID: 37296475
- PMCID: PMC10257282
- DOI: 10.1186/s13256-023-03984-0
Terminal microdeletion of chromosome 18 in a Malaysian boy characterized with few features of typical 18q- deletion syndrome: a case report
Abstract
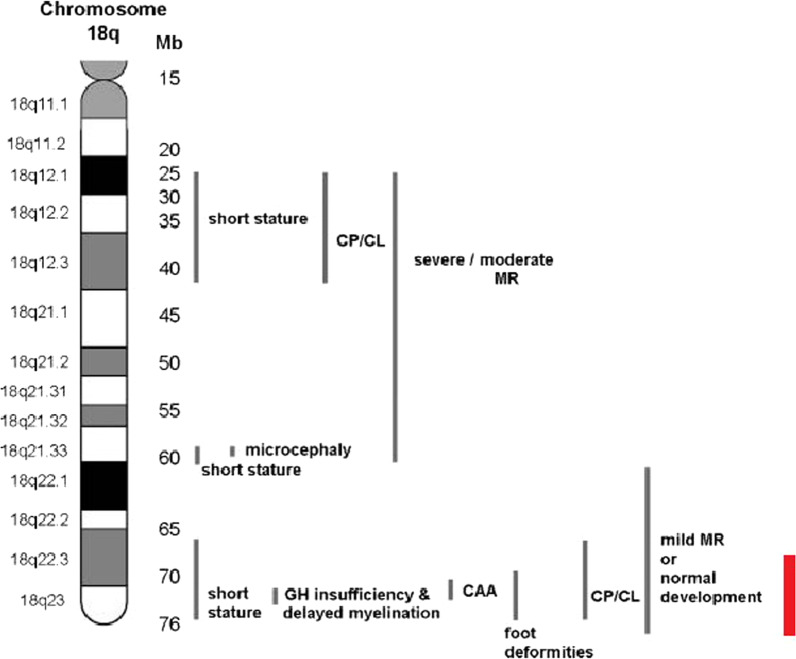
Background: The 18q- deletion syndrome is a rare congenital chromosomal disorder caused by a partial deletion of the long arm of chromosome 18. The diagnosis of a patient with this syndrome relies on the family medical history, physical examination, developmental assessment, and cytogenetic findings. However, the phenotype of patients with 18q- deletion syndrome can be highly variable, ranging from almost normal to severe malformations and intellectual disability, and normal cytogenetic findings are common, thus complicating the diagnosis. Interestingly, only few characteristic features of typical 18q- deletion syndrome were found in the patient, despite sharing the same critical region. To our knowledge, this is the first report of a Malaysian individual with 18q- terminal microdeletion diagnosed with microarray-based technology.
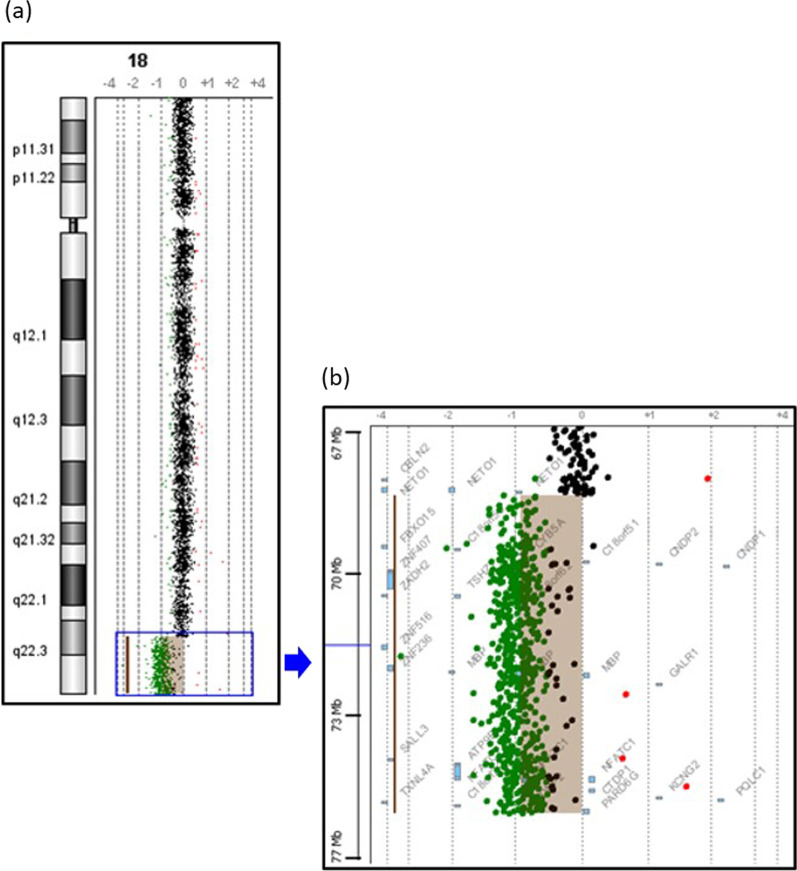
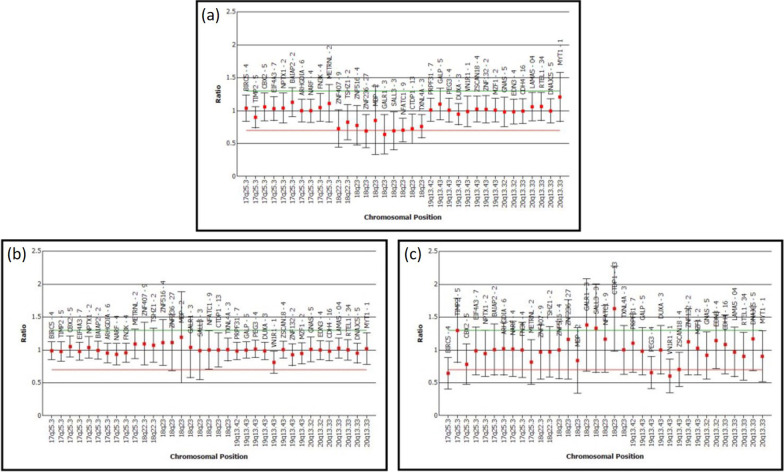
Case presentation: Here we report a 16-year-old Malaysian Chinese boy, a product of a non-consanguineous marriage, who presented with intellectual disability, facial dysmorphism, high arched palate, congenital talipes equinovarus (clubfoot), congenital scoliosis, congenital heart defect, and behavioral problems. A routine chromosome analysis on 20 metaphase cells showed a normal 46, XY G-banded karyotype. Array-based comparative genomic hybridization was performed using a commercially available 244 K 60-mer oligonucleotide microarray slide according to the manufacturer's protocol. This platform allows genome-wide survey and molecular profiling of genomic aberrations with an average resolution of about 10 kB. In addition, multiplex ligation-dependent probe amplification analysis was carried out using SALSA MLPA kit P320 Telomere-13 to confirm the array-based comparative genomic hybridization finding. Array-based comparative genomic hybridization analysis revealed a 7.3 MB terminal deletion involving chromosome band 18q22.3-qter. This finding was confirmed by multiplex ligation-dependent probe amplification, where a deletion of ten probes mapping to the 18q22.3-q23 region was detected, and further multiplex ligation-dependent probe amplification analysis on his parents showed the deletion to be de novo.
Conclusion: The findings from this study expand the phenotypic spectrum of the 18q- deletion syndrome by presenting a variation of typical 18q- deletion syndrome features to the literature. In addition, this case report demonstrated the ability of the molecular karyotyping method, such as array-based comparative genomic hybridization, to assist in the diagnosis of cases with a highly variable phenotype and variable aberrations, such as 18q- deletion syndrome.
Keywords: 18q- deletion syndrome; Array-CGH; Congenital chromosomal disorder; Dysmorphism; Intellectual disability.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
