

HLA-II immunopeptidome profiling and deep learning reveal features of antigenicity to inform antigen discovery
- PMID: 37301199
- PMCID: PMC10519123
- DOI: 10.1016/j.immuni.2023.05.009
HLA-II immunopeptidome profiling and deep learning reveal features of antigenicity to inform antigen discovery
Abstract
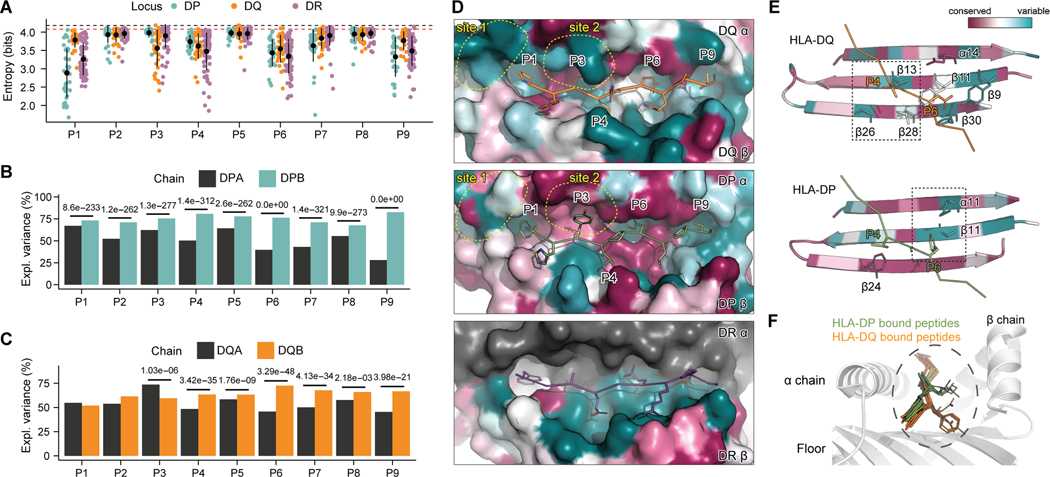
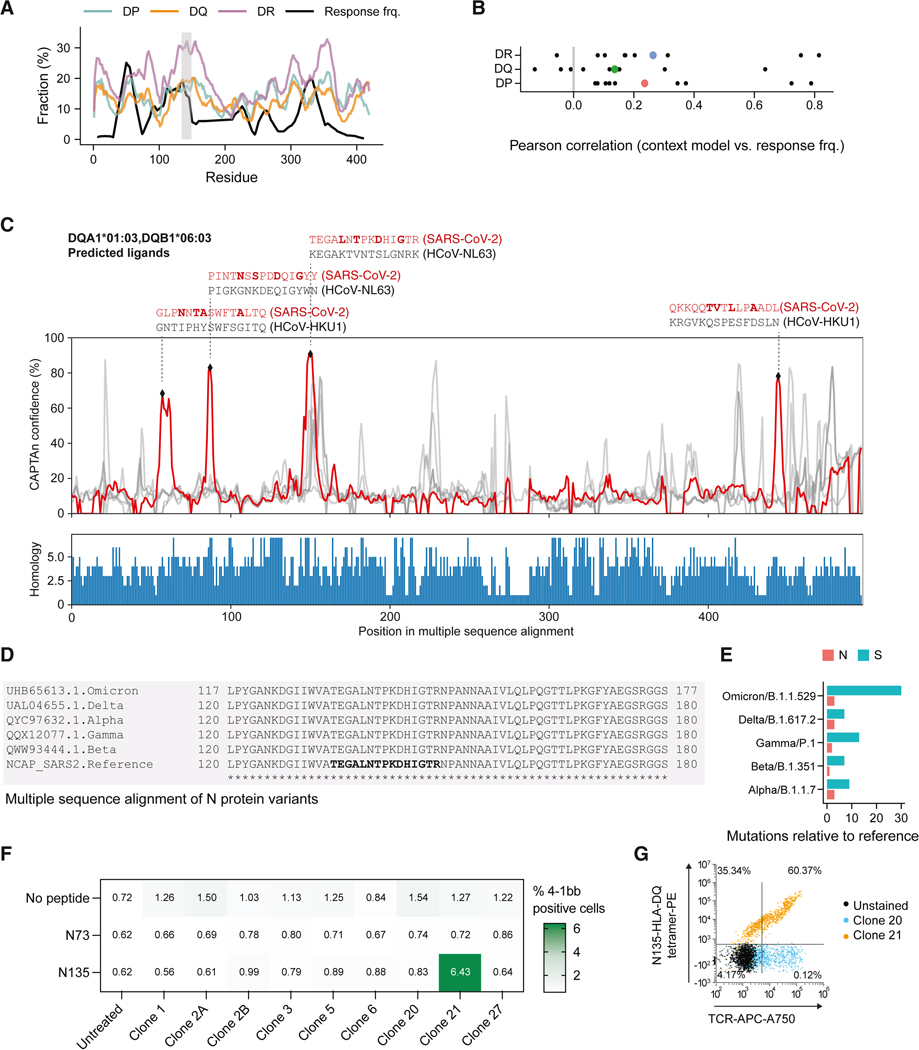
CD4+ T cell responses are exquisitely antigen specific and directed toward peptide epitopes displayed by human leukocyte antigen class II (HLA-II) on antigen-presenting cells. Underrepresentation of diverse alleles in ligand databases and an incomplete understanding of factors affecting antigen presentation in vivo have limited progress in defining principles of peptide immunogenicity. Here, we employed monoallelic immunopeptidomics to identify 358,024 HLA-II binders, with a particular focus on HLA-DQ and HLA-DP. We uncovered peptide-binding patterns across a spectrum of binding affinities and enrichment of structural antigen features. These aspects underpinned the development of context-aware predictor of T cell antigens (CAPTAn), a deep learning model that predicts peptide antigens based on their affinity to HLA-II and full sequence of their source proteins. CAPTAn was instrumental in discovering prevalent T cell epitopes from bacteria in the human microbiome and a pan-variant epitope from SARS-CoV-2. Together CAPTAn and associated datasets present a resource for antigen discovery and the unraveling genetic associations of HLA alleles with immunopathologies.
Keywords: CD4(+) T cells; MHC class II; SARS-CoV-2 antigens; antigen presentation; immunopeptidomics; microbiome antigens; protein sequence models.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.J.X. is a co-founder of Celsius Therapeutics and Jnana Therapeutics, a member of the Scientific Advisory Board at Nestle, and a member of the Board of Directors at Moonlake Immunotherapeutics. S.A.C. is a member of the scientific advisory boards of Kymera, PTM BioLabs, Seer, and PrognomIQ.
Figures





References
-
- Borst J, Ahrends T, Bąbała N, Melief CJM, and Kastenmüller W. (2018). CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol 18, 635–647. - PubMed
-
- Germain RN, and Margulies DH (1993). The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol 11, 403–450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
