

Extreme phenotypic heterogeneity in non-expansion spinocerebellar ataxias
- PMID: 37301203
- PMCID: PMC10357418
- DOI: 10.1016/j.ajhg.2023.05.009
Extreme phenotypic heterogeneity in non-expansion spinocerebellar ataxias
Abstract
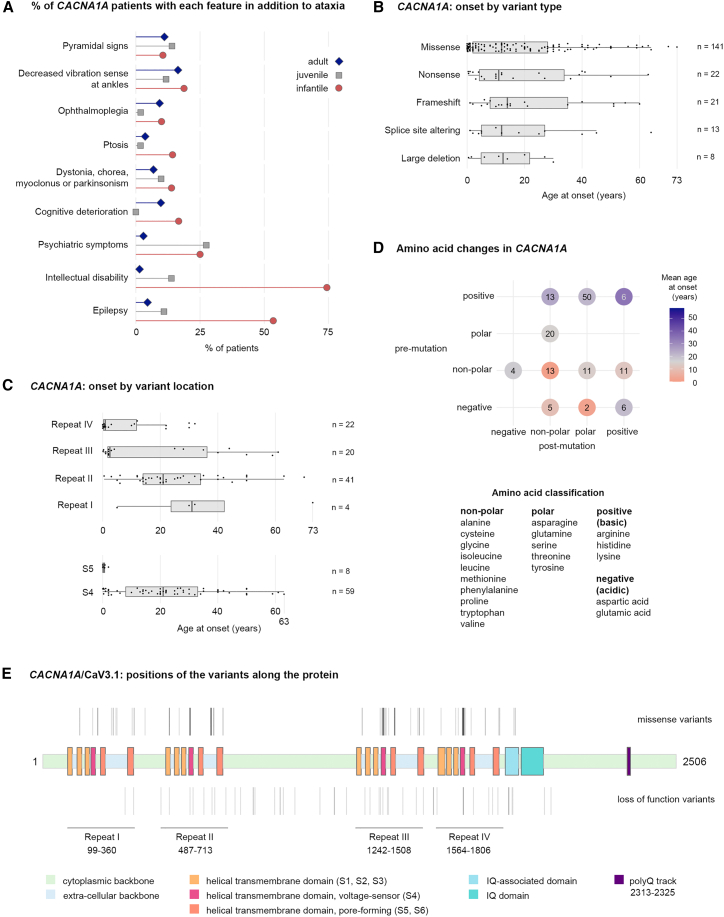
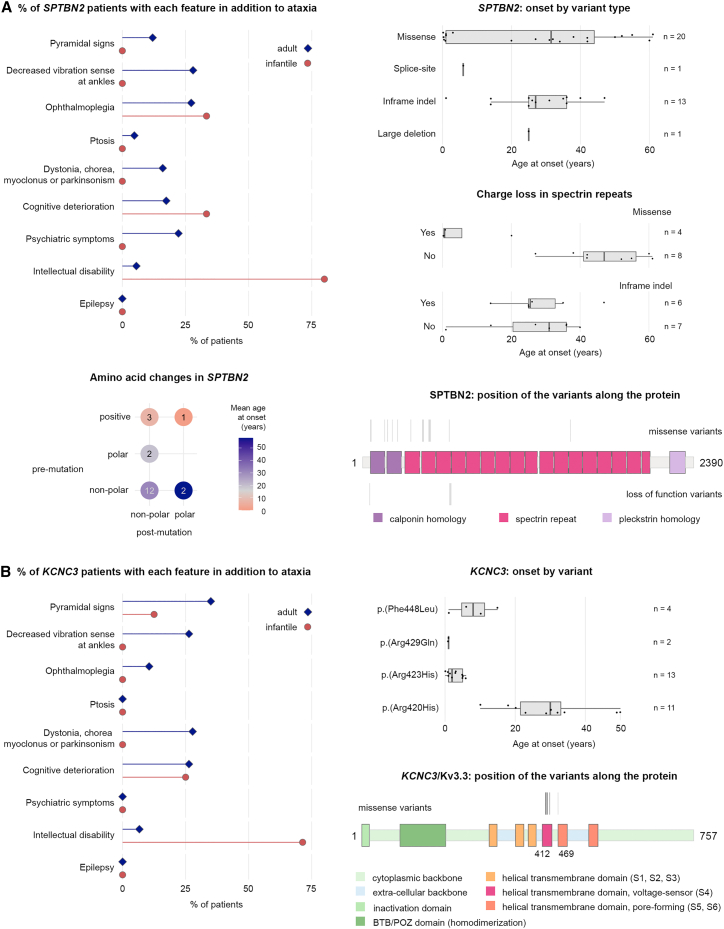
Although the best-known spinocerebellar ataxias (SCAs) are triplet repeat diseases, many SCAs are not caused by repeat expansions. The rarity of individual non-expansion SCAs, however, has made it difficult to discern genotype-phenotype correlations. We therefore screened individuals who had been found to bear variants in a non-expansion SCA-associated gene through genetic testing, and after we eliminated genetic groups that had fewer than 30 subjects, there were 756 subjects bearing single-nucleotide variants or deletions in one of seven genes: CACNA1A (239 subjects), PRKCG (175), AFG3L2 (101), ITPR1 (91), STUB1 (77), SPTBN2 (39), or KCNC3 (34). We compared age at onset, disease features, and progression by gene and variant. There were no features that reliably distinguished one of these SCAs from another, and several genes-CACNA1A, ITPR1, SPTBN2, and KCNC3-were associated with both adult-onset and infantile-onset forms of disease, which also differed in presentation. Nevertheless, progression was overall very slow, and STUB1-associated disease was the fastest. Several variants in CACNA1A showed particularly wide ranges in age at onset: one variant produced anything from infantile developmental delay to ataxia onset at 64 years of age within the same family. For CACNA1A, ITPR1, and SPTBN2, the type of variant and charge change on the protein greatly affected the phenotype, defying pathogenicity prediction algorithms. Even with next-generation sequencing, accurate diagnosis requires dialogue between the clinician and the geneticist.
Keywords: Spinocerebellar Ataxia, SCA, CACNA1A, PRKCG, AFG3L2, ITPR1, STUB1, SPTBN2, KCNC3, onset.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interest.
Figures



References
-
- Klockgether T., Mariotti C., Paulson H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers. 2019;5:24. - PubMed
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. - PubMed
-
- Ruano L., Melo C., Silva M.C., Coutinho P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology. 2014;42:174–183. - PubMed
-
- Nibbeling E.A.R., Duarri A., Verschuuren-Bemelmans C.C., Fokkens M.R., Karjalainen J.M., Smeets C.J.L.M., de Boer-Bergsma J.J., van der Vries G., Dooijes D., Bampi G.B., et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain. 2017;140:2860–2878. - PubMed
-
- Jodice C., Mantuano E., Veneziano L., Trettel F., Sabbadini G., Calandriello L., Francia A., Spadaro M., Pierelli F., Salvi F., et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum. Mol. Genet. 1997;6:1973–1978. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
