

Impaired histone inheritance promotes tumor progression
- PMID: 37301892
- PMCID: PMC10257670
- DOI: 10.1038/s41467-023-39185-y
Impaired histone inheritance promotes tumor progression
Abstract
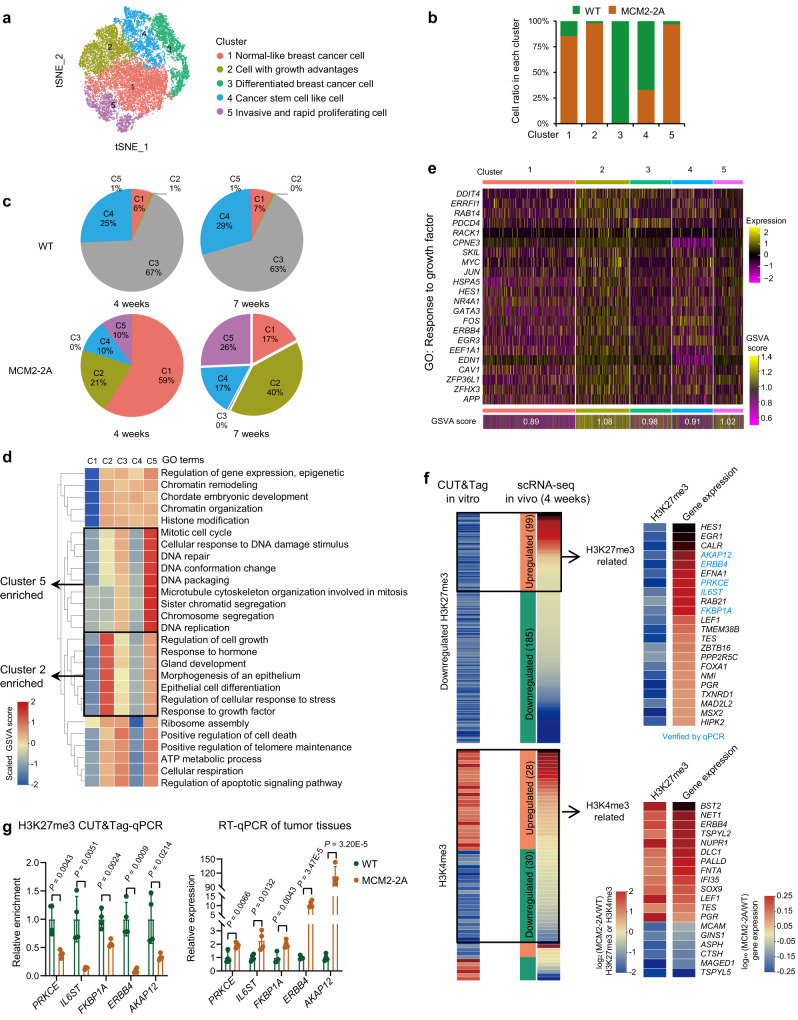
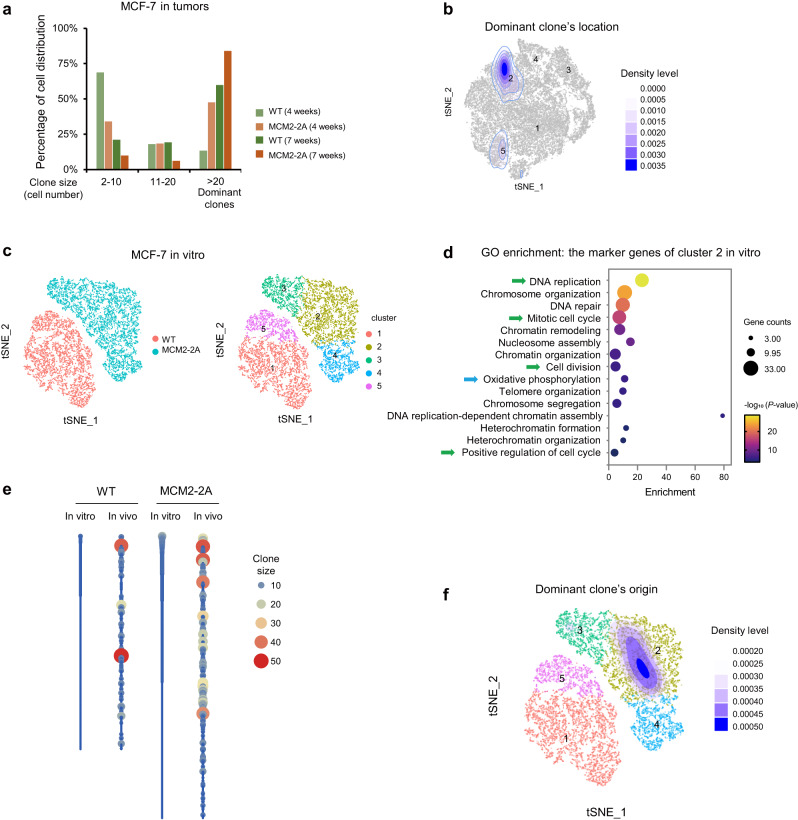
Faithful inheritance of parental histones is essential to maintain epigenetic information and cellular identity during cell division. Parental histones are evenly deposited onto the replicating DNA of sister chromatids in a process dependent on the MCM2 subunit of DNA helicase. However, the impact of aberrant parental histone partition on human disease such as cancer is largely unknown. In this study, we construct a model of impaired histone inheritance by introducing MCM2-2A mutation (defective in parental histone binding) in MCF-7 breast cancer cells. The resulting impaired histone inheritance reprograms the histone modification landscapes of progeny cells, especially the repressive histone mark H3K27me3. Lower H3K27me3 levels derepress the expression of genes associated with development, cell proliferation, and epithelial to mesenchymal transition. These epigenetic changes confer fitness advantages to some newly emerged subclones and consequently promote tumor growth and metastasis after orthotopic implantation. In summary, our results indicate that impaired inheritance of parental histones can drive tumor progression.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
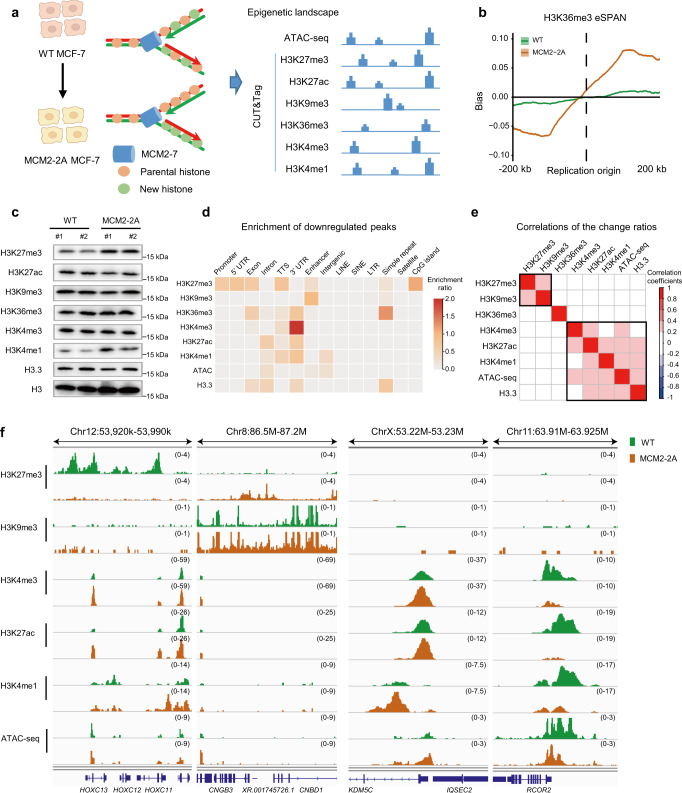
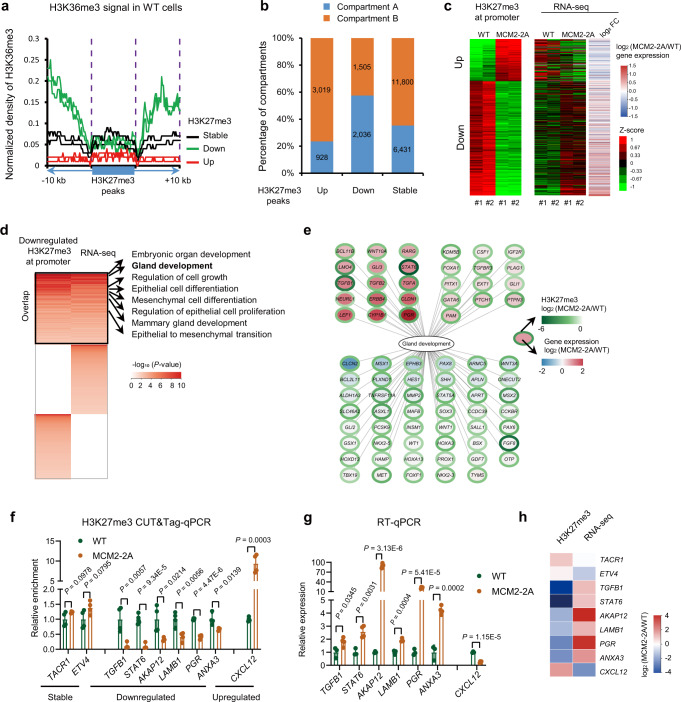
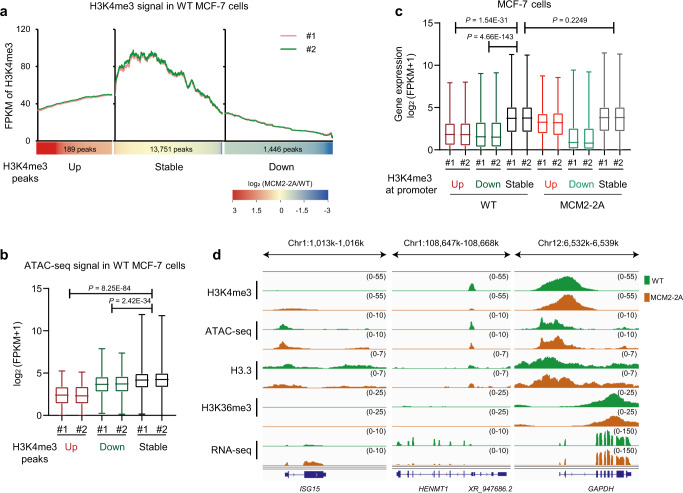
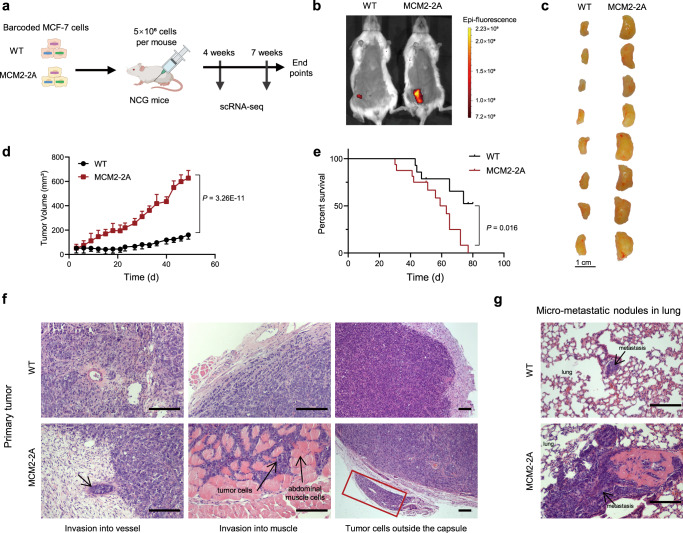
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
