

Implementation of Nanopore sequencing as a pragmatic workflow for copy number variant confirmation in the clinic
- PMID: 37301971
- PMCID: PMC10257846
- DOI: 10.1186/s12967-023-04243-y
Implementation of Nanopore sequencing as a pragmatic workflow for copy number variant confirmation in the clinic
Abstract
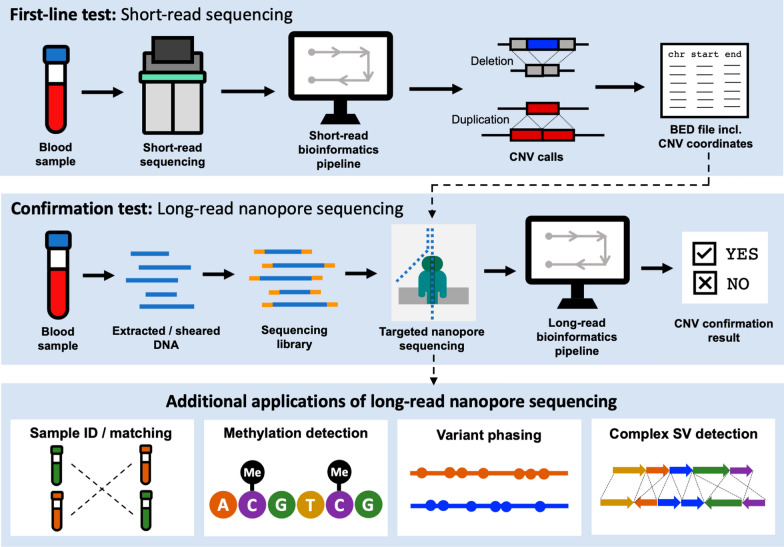
Background: Diagnosis of rare genetic diseases can be a long, expensive and complex process, involving an array of tests in the hope of obtaining an actionable result. Long-read sequencing platforms offer the opportunity to make definitive molecular diagnoses using a single assay capable of detecting variants, characterizing methylation patterns, resolving complex rearrangements, and assigning findings to long-range haplotypes. Here, we demonstrate the clinical utility of Nanopore long-read sequencing by validating a confirmatory test for copy number variants (CNVs) in neurodevelopmental disorders and illustrate the broader applications of this platform to assess genomic features with significant clinical implications.
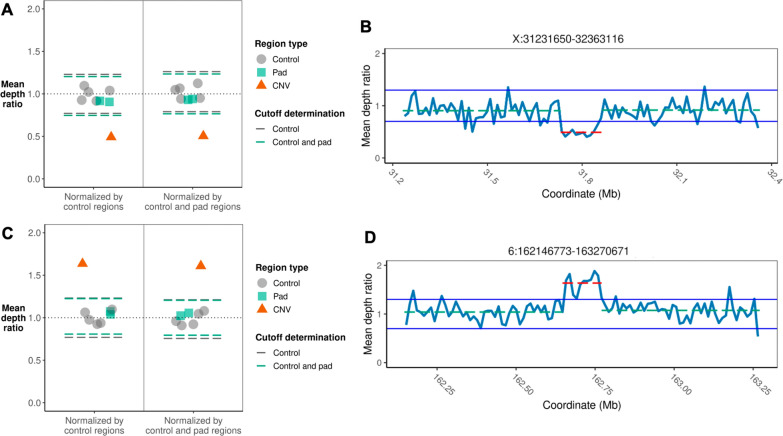
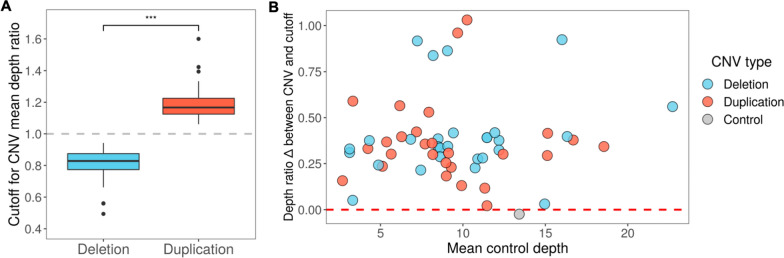
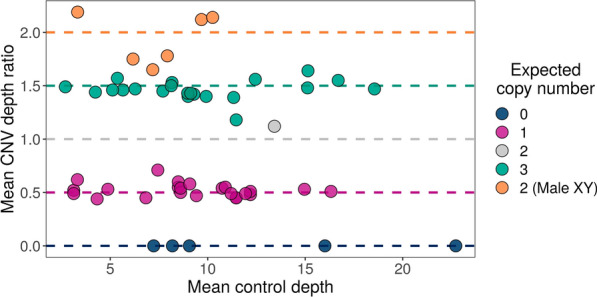
Methods: We used adaptive sampling on the Oxford Nanopore platform to sequence 25 genomic DNA samples and 5 blood samples collected from patients with known or false-positive copy number changes originally detected using short-read sequencing. Across the 30 samples (a total of 50 with replicates), we assayed 35 known unique CNVs (a total of 55 with replicates) and one false-positive CNV, ranging in size from 40 kb to 155 Mb, and assessed the presence or absence of suspected CNVs using normalized read depth.
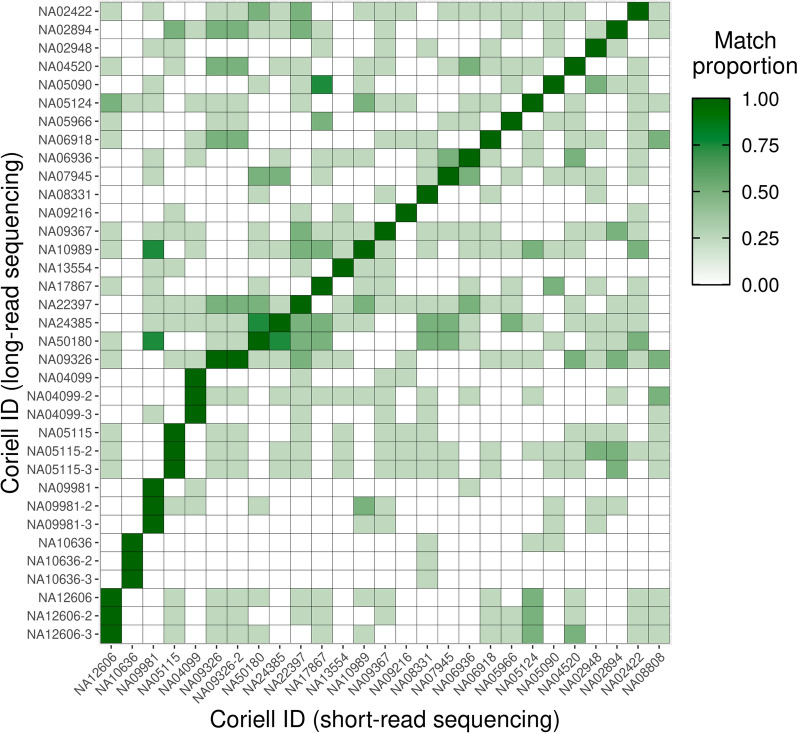
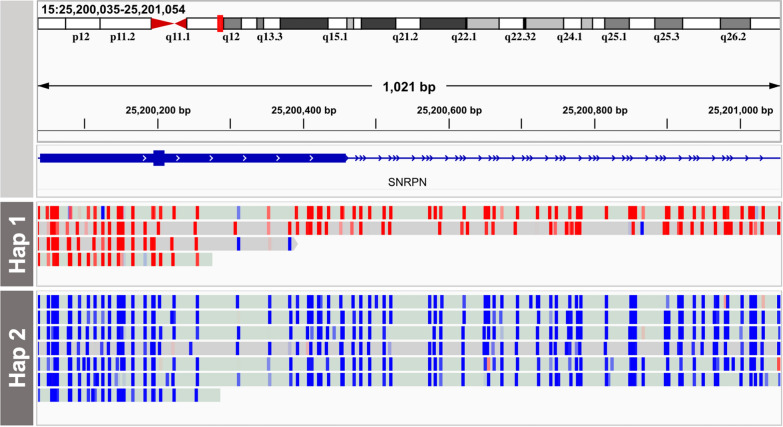
Results: Across 50 samples (including replicates) sequenced on individual MinION flow cells, we achieved an average on-target mean depth of 9.5X and an average on-target read length of 4805 bp. Using a custom read depth-based analysis, we successfully confirmed the presence of all 55 known CNVs (including replicates) and the absence of one false-positive CNV. Using the same CNV-targeted data, we compared genotypes of single nucleotide variant loci to verify that no sample mix-ups occurred between assays. For one case, we also used methylation detection and phasing to investigate the parental origin of a 15q11.2-q13 duplication with implications for clinical prognosis.
Conclusions: We present an assay that efficiently targets genomic regions to confirm clinically relevant CNVs with a concordance rate of 100%. Furthermore, we demonstrate how integration of genotype, methylation, and phasing data from the Nanopore sequencing platform can potentially simplify and shorten the diagnostic odyssey.
Keywords: Adaptive sampling; Clinical testing; Copy number variants; Genome analysis; Long-read sequencing; Neurodevelopmental disorders; Oxford Nanopore Technologies; Targeted sequencing.
© 2023. The Author(s).
Conflict of interest statement
S.U.G, J.B., D.H., P.S., M.R., K.I., and A.K. are either current or previous employees of MyOme, and B.L. and D.E.M. are either current or previous consultants with MyOme. D.E.M is engaged in a research agreement with Oxford Nanopore, and they have paid for him to travel to speak on their behalf. A provisional patent application has been filed on the route of sample identification described in this study.
Figures
Similar articles
-
Oxford Nanopore long-read sequencing with CRISPR/Cas9-mediated target selection for accurate characterization of copy number variants in the LDLR gene.Eur J Med Genet. 2025 Apr;74:105003. doi: 10.1016/j.ejmg.2025.105003. Epub 2025 Feb 22. Eur J Med Genet. 2025. PMID: 39993709
-
A Comprehensive Workflow for Read Depth-Based Identification of Copy-Number Variation from Whole-Genome Sequence Data.Am J Hum Genet. 2018 Jan 4;102(1):142-155. doi: 10.1016/j.ajhg.2017.12.007. Am J Hum Genet. 2018. PMID: 29304372 Free PMC article.
-
Preclinical workup using long-read amplicon sequencing provides families with de novo pathogenic variants access to universal preimplantation genetic testing.Hum Reprod. 2023 Mar 1;38(3):511-519. doi: 10.1093/humrep/deac273. Hum Reprod. 2023. PMID: 36625546
-
Best Practices in Microbial Experimental Evolution: Using Reporters and Long-Read Sequencing to Identify Copy Number Variation in Experimental Evolution.J Mol Evol. 2023 Jun;91(3):356-368. doi: 10.1007/s00239-023-10102-7. Epub 2023 Apr 3. J Mol Evol. 2023. PMID: 37012421 Free PMC article. Review.
-
Free-access copy-number variant detection tools for targeted next-generation sequencing data.Mutat Res Rev Mutat Res. 2019 Jan-Mar;779:114-125. doi: 10.1016/j.mrrev.2019.02.005. Epub 2019 Feb 23. Mutat Res Rev Mutat Res. 2019. PMID: 31097148 Review.
Cited by
-
De novel heterozygous copy number deletion on 7q31.31-7q31.32 involving TSPAN12 gene with familial exudative vitreoretinopathy in a Chinese family.Int J Ophthalmol. 2023 Dec 18;16(12):1952-1961. doi: 10.18240/ijo.2023.12.06. eCollection 2023. Int J Ophthalmol. 2023. PMID: 38111929 Free PMC article.
-
Concordance of Whole-Genome Long-Read Sequencing with Standard Clinical Testing for Prader-Willi and Angelman Syndromes.J Mol Diagn. 2025 Mar;27(3):166-176. doi: 10.1016/j.jmoldx.2024.12.003. Epub 2025 Jan 3. J Mol Diagn. 2025. PMID: 39756651
-
Haplotypic resolution of the challenging genomic regions of MHC and KIR using a combination of targeted sequencing and a novel assembly pipeline.Nucleic Acids Res. 2025 May 22;53(10):gkaf441. doi: 10.1093/nar/gkaf441. Nucleic Acids Res. 2025. PMID: 40464686 Free PMC article.
-
Benchmarking nanopore sequencing and rapid genomics feasibility: validation at a quaternary hospital in New Zealand.NPJ Genom Med. 2024 Nov 8;9(1):57. doi: 10.1038/s41525-024-00445-5. NPJ Genom Med. 2024. PMID: 39516456 Free PMC article.
-
Applying the National Genomic DNA Reference Materials to Evaluate the Performance of Nanopore Sequencing in Identifying Thalassemia Variants.J Clin Lab Anal. 2025 Jun;39(11):e70044. doi: 10.1002/jcla.70044. Epub 2025 May 20. J Clin Lab Anal. 2025. PMID: 40394932 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
