

Peripheral Fragile X messenger ribonucleoprotein is required for the timely closure of a critical period for neuronal susceptibility in the ventral cochlear nucleus
- PMID: 37305436
- PMCID: PMC10248243
- DOI: 10.3389/fncel.2023.1186630
Peripheral Fragile X messenger ribonucleoprotein is required for the timely closure of a critical period for neuronal susceptibility in the ventral cochlear nucleus
Abstract
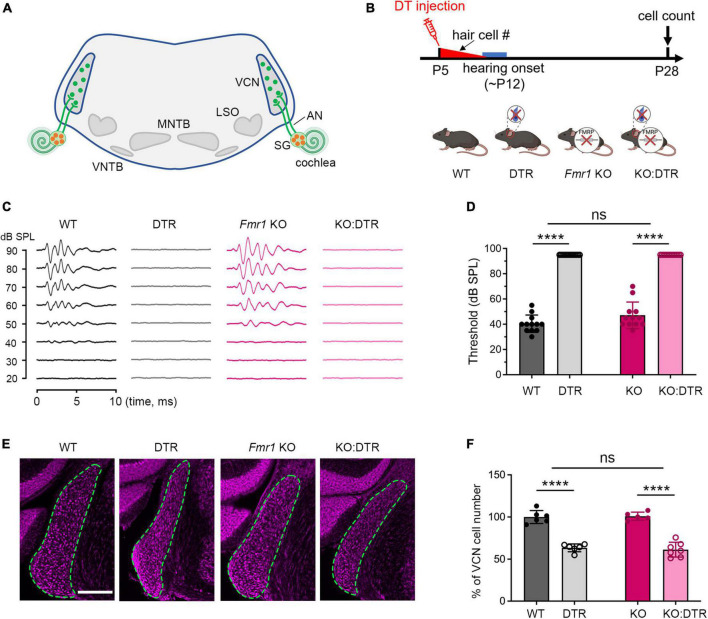
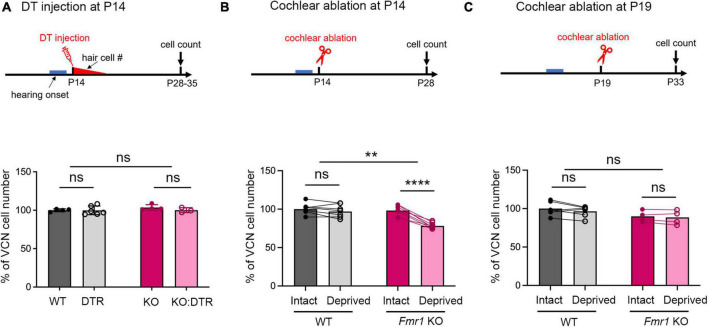
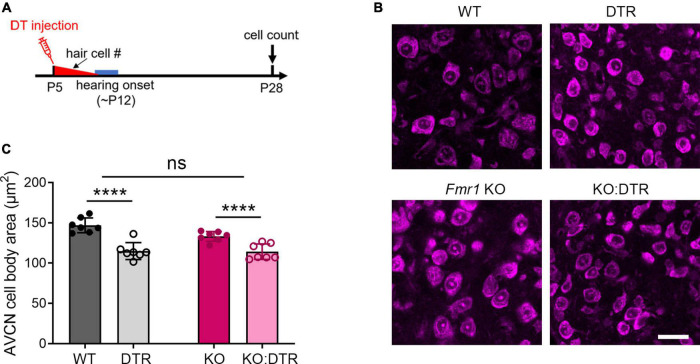
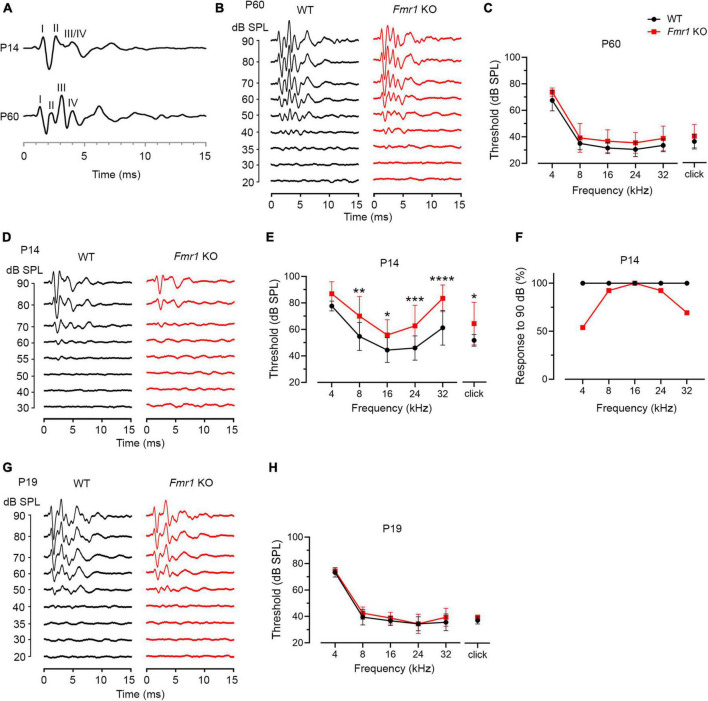
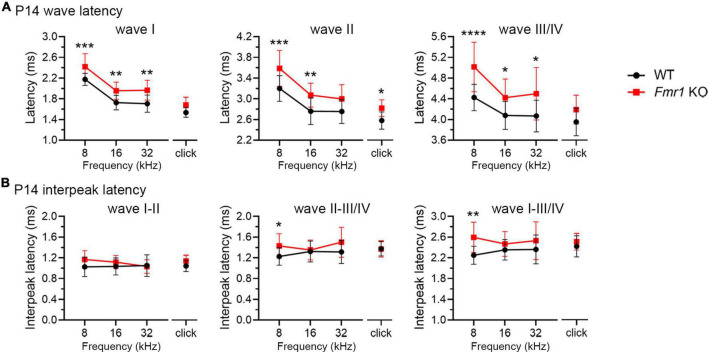
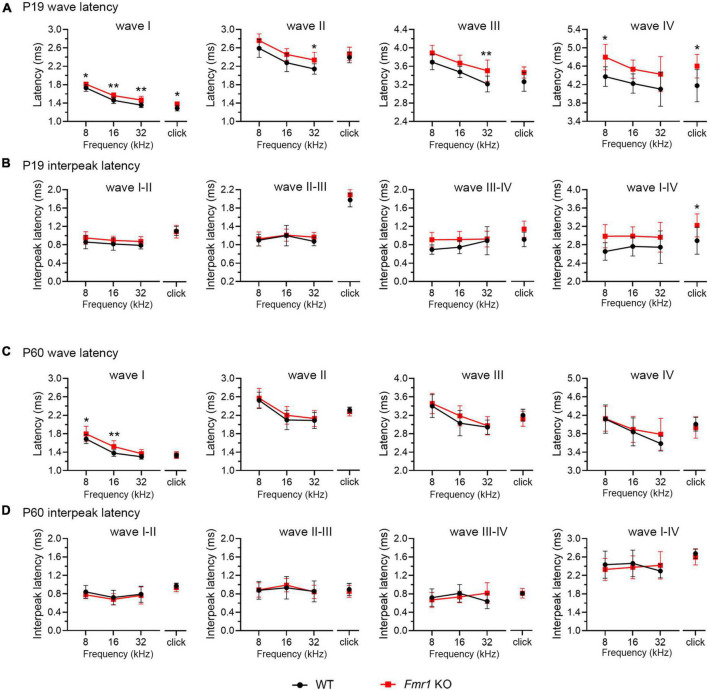
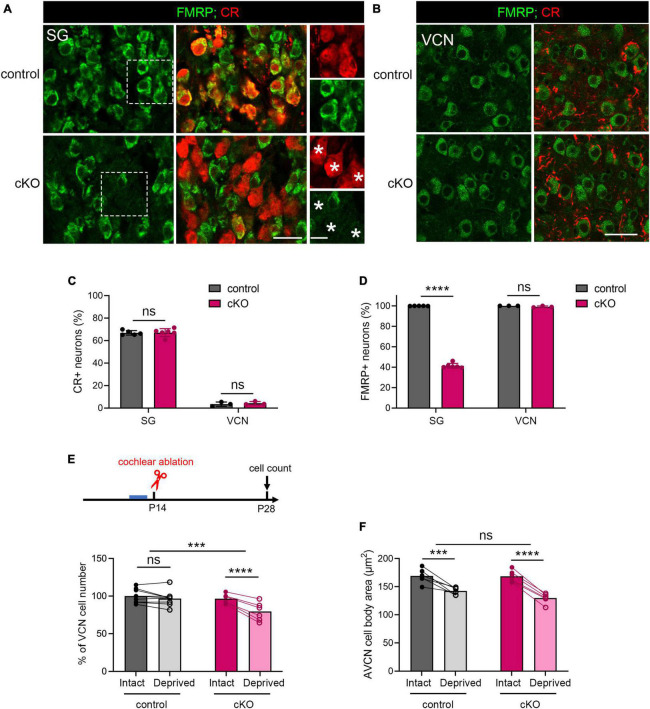
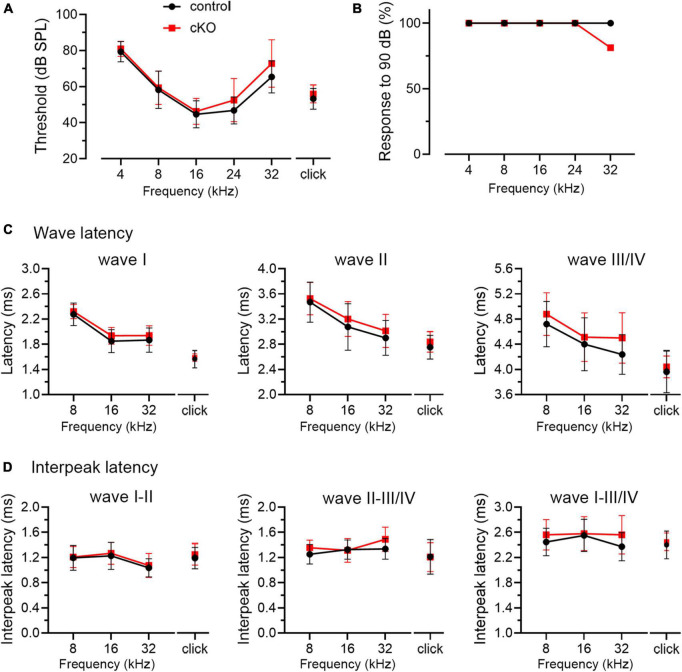
Alterations in neuronal plasticity and critical periods are common across neurodevelopmental diseases, including Fragile X syndrome (FXS), the leading single-gene cause of autism. Characterized with sensory dysfunction, FXS is the result of gene silencing of Fragile X messenger ribonucleoprotein 1 (FMR1) and loss of its product, Fragile X messenger ribonucleoprotein (FMRP). The mechanisms underlying altered critical period and sensory dysfunction in FXS are obscure. Here, we performed genetic and surgical deprivation of peripheral auditory inputs in wildtype and Fmr1 knockout (KO) mice across ages and investigated the effects of global FMRP loss on deafferentation-induced neuronal changes in the ventral cochlear nucleus (VCN) and auditory brainstem responses. The degree of neuronal cell loss during the critical period was unchanged in Fmr1 KO mice. However, the closure of the critical period was delayed. Importantly, this delay was temporally coincidental with reduced hearing sensitivity, implying an association with sensory inputs. Functional analyses further identified early-onset and long-lasting alterations in signal transmission from the spiral ganglion to the VCN, suggesting a peripheral site of FMRP action. Finally, we generated conditional Fmr1 KO (cKO) mice with selective deletion of FMRP in spiral ganglion but not VCN neurons. cKO mice recapitulated the delay in the VCN critical period closure in Fmr1 KO mice, confirming an involvement of cochlear FMRP in shaping the temporal features of neuronal critical periods in the brain. Together, these results identify a novel peripheral mechanism of neurodevelopmental pathogenesis.
Keywords: Fragile X syndrome; afferent influence; brain development; critical period; hearing onset; sensory organ.
Copyright © 2023 Yu and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Selective Deletion of Astroglial FMRP Dysregulates Glutamate Transporter GLT1 and Contributes to Fragile X Syndrome Phenotypes In Vivo.J Neurosci. 2016 Jul 6;36(27):7079-94. doi: 10.1523/JNEUROSCI.1069-16.2016. J Neurosci. 2016. PMID: 27383586 Free PMC article.
-
Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome.Neurobiol Dis. 2016 May;89:126-35. doi: 10.1016/j.nbd.2016.02.002. Epub 2016 Feb 2. Neurobiol Dis. 2016. PMID: 26850918 Free PMC article.
-
Abnormal neuronal morphology and neurochemistry in the auditory brainstem of Fmr1 knockout rats.Neuroscience. 2015 Sep 10;303:285-98. doi: 10.1016/j.neuroscience.2015.06.061. Epub 2015 Jul 9. Neuroscience. 2015. PMID: 26166728
-
Modeling fragile X syndrome in the Fmr1 knockout mouse.Intractable Rare Dis Res. 2014 Nov;3(4):118-33. doi: 10.5582/irdr.2014.01024. Intractable Rare Dis Res. 2014. PMID: 25606362 Free PMC article. Review.
-
BDNF in fragile X syndrome.Neuropharmacology. 2014 Jan;76 Pt C:729-36. doi: 10.1016/j.neuropharm.2013.05.018. Epub 2013 May 29. Neuropharmacology. 2014. PMID: 23727436 Review.
Cited by
-
The Development of Speaking and Singing in Infants May Play a Role in Genomics and Dementia in Humans.Brain Sci. 2023 Aug 11;13(8):1190. doi: 10.3390/brainsci13081190. Brain Sci. 2023. PMID: 37626546 Free PMC article. Review.
-
Atypical Retinal Ganglion Cell Function in a Mouse Model of Fragile X Syndrome.J Neurosci. 2025 Jul 2;45(27):e0013252025. doi: 10.1523/JNEUROSCI.0013-25.2025. J Neurosci. 2025. PMID: 40467301 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials