

Temozolomide Sensitizes ARID1A-Mutated Cancers to PARP Inhibitors
- PMID: 37306706
- PMCID: PMC10527942
- DOI: 10.1158/0008-5472.CAN-22-3646
Temozolomide Sensitizes ARID1A-Mutated Cancers to PARP Inhibitors
Abstract
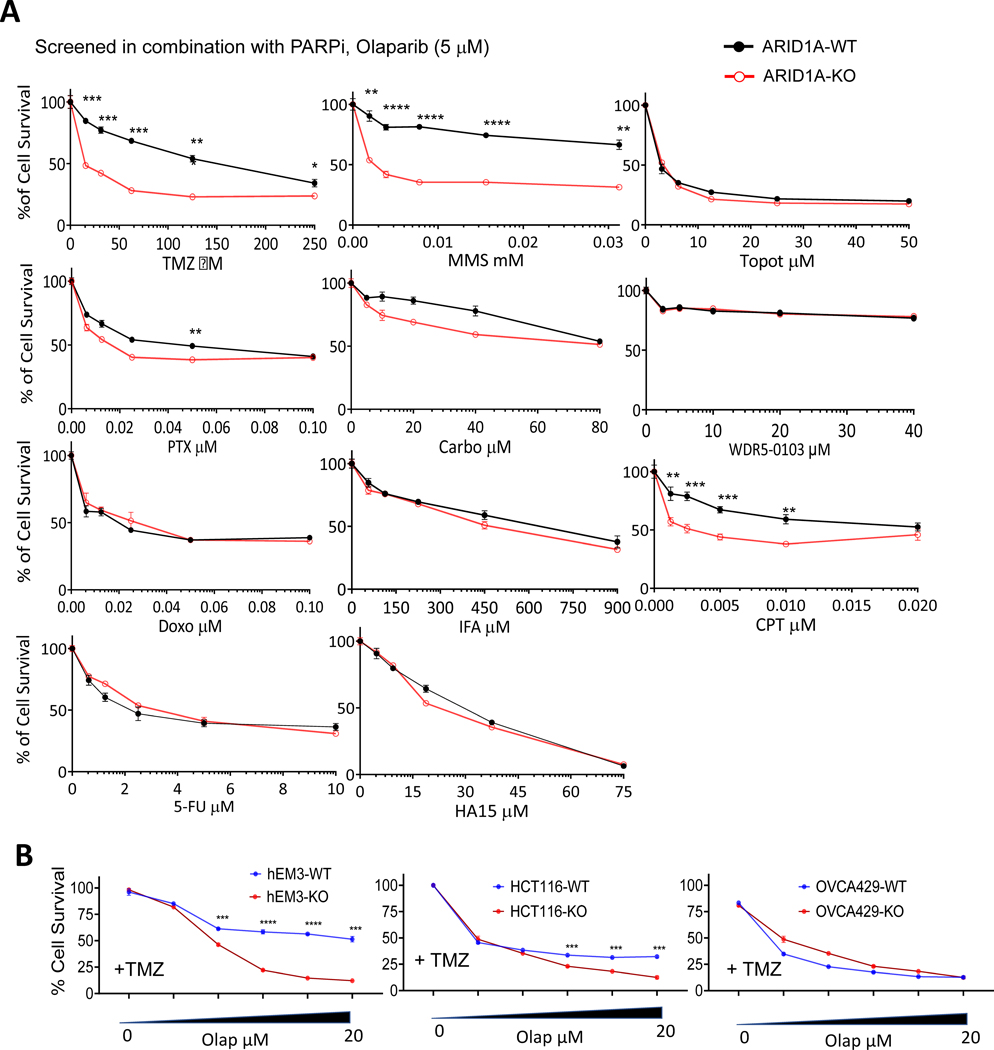
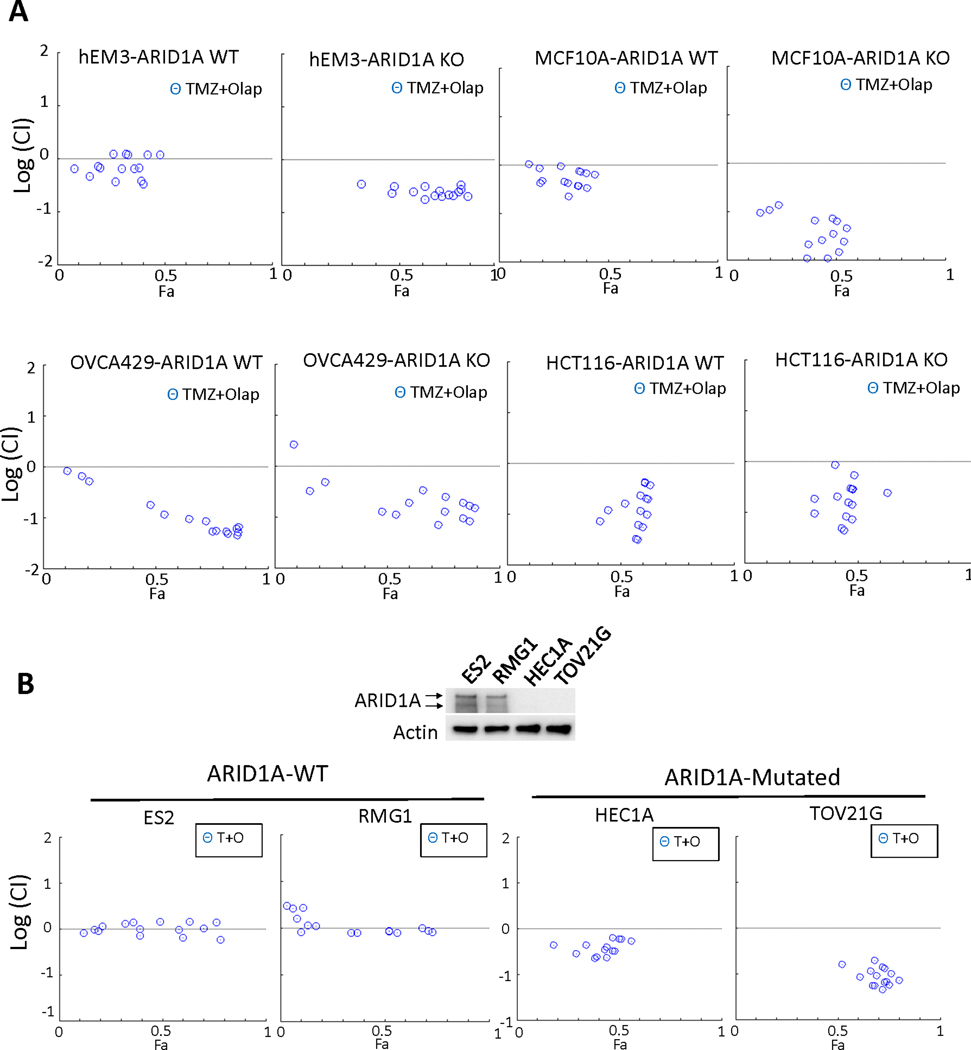
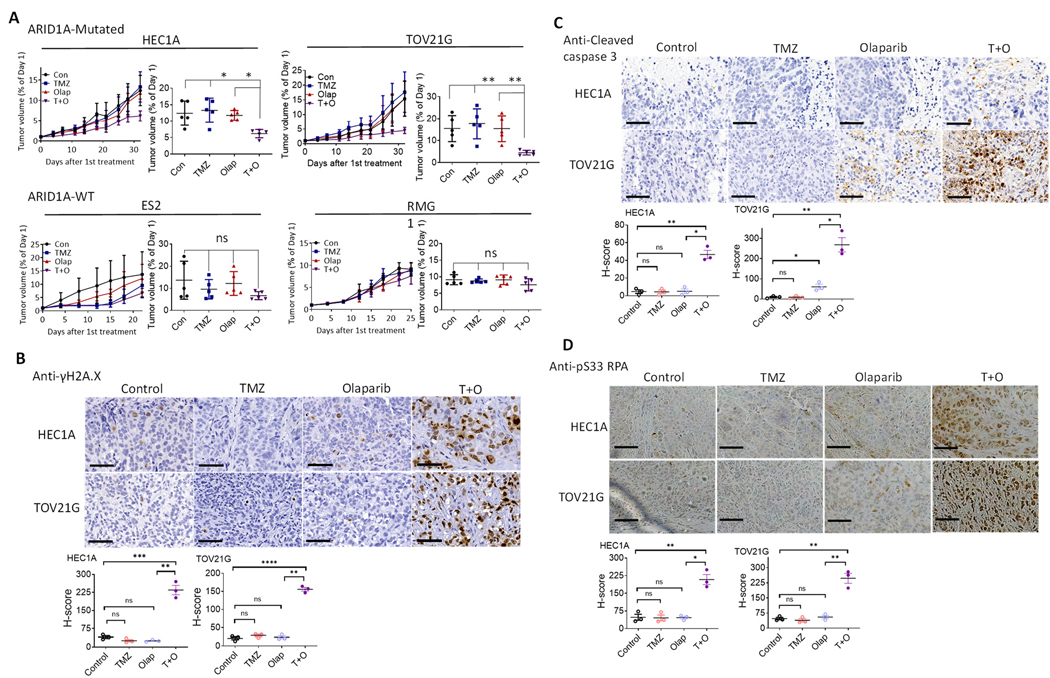
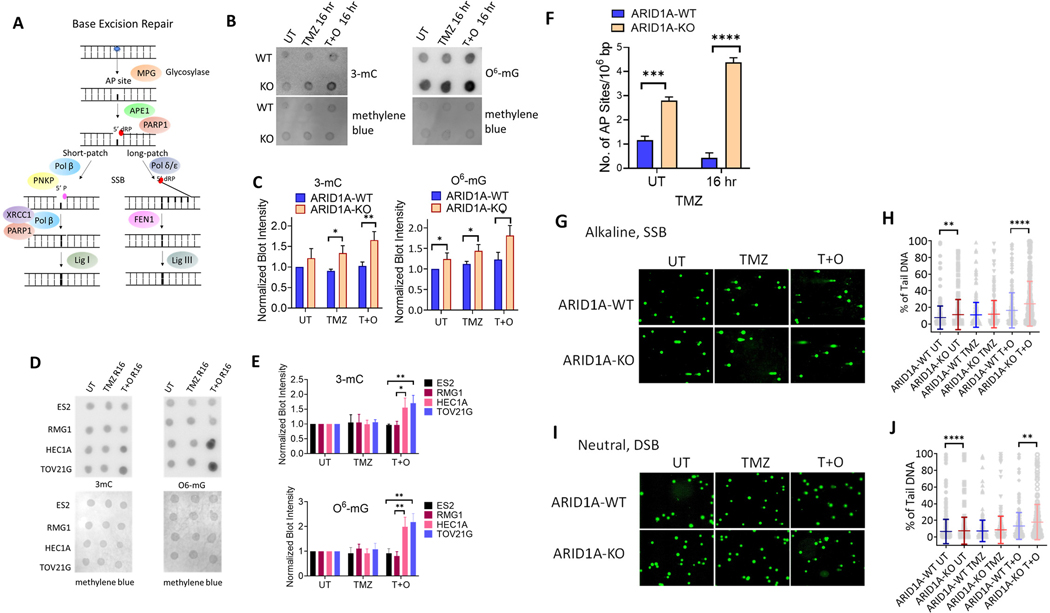
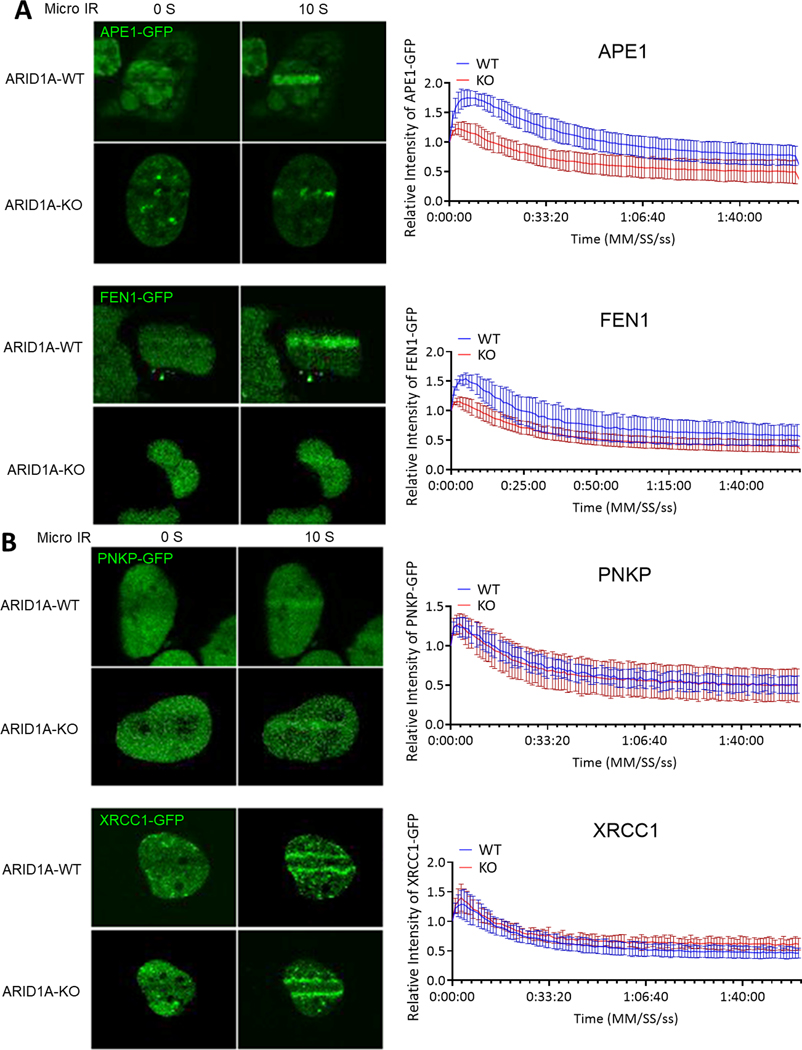
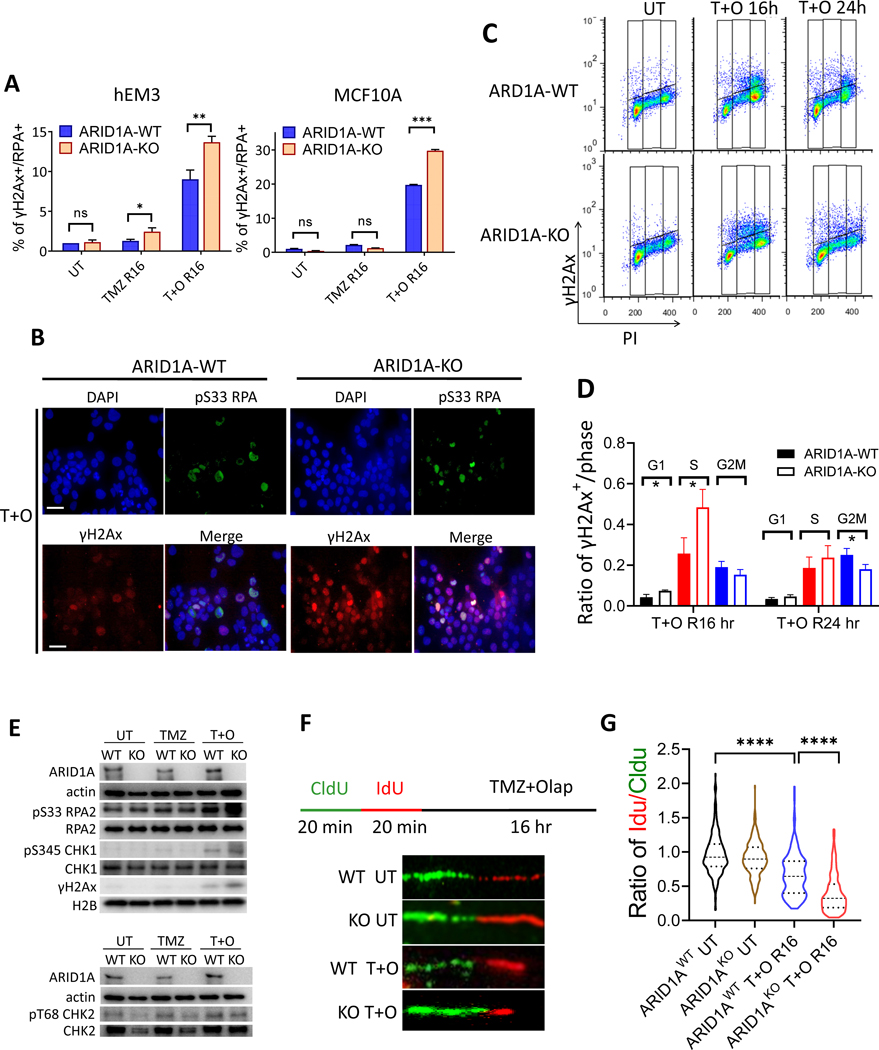
ARID1A is a subunit of SWI/SNF chromatin remodeling complexes and is mutated in many types of human cancers, especially those derived from endometrial epithelium, including ovarian and uterine clear cell carcinoma (CCC) and endometrioid carcinoma (EMCA). Loss-of-function mutations in ARID1A alter epigenetic regulation of transcription, cell-cycle checkpoint control, and DNA damage repair. We report here that mammalian cells with ARID1A deficiency harbor accumulated DNA base lesions and increased abasic (AP) sites, products of glycosylase in the first step of base excision repair (BER). ARID1A mutations also delayed recruitment kinetics of BER long-patch repair effectors. Although ARID1A-deficient tumors were not sensitive to monotherapy with DNA-methylating temozolomide (TMZ), the combination of TMZ with PARP inhibitors (PARPi) potently elicited double-strand DNA breaks, replication stress, and replication fork instability in ARID1A-deficient cells. The TMZ and PARPi combination also significantly delayed in vivo growth of ovarian tumor xenografts carrying ARID1A mutations and induced apoptosis and replication stress in xenograft tumors. Together, these findings identified a synthetic lethal strategy to enhance the response of ARID1A-mutated cancers to PARP inhibition, which warrants further experimental exploration and clinical trial validation.
Significance: The combination of temozolomide and PARP inhibitor exploits the specific DNA damage repair status of ARID1A-inactivated ovarian cancers to suppress tumor growth.
©2023 American Association for Cancer Research.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
