

Characterization of p38α autophosphorylation inhibitors that target the non-canonical activation pathway
- PMID: 37308482
- PMCID: PMC10261013
- DOI: 10.1038/s41467-023-39051-x
Characterization of p38α autophosphorylation inhibitors that target the non-canonical activation pathway
Abstract
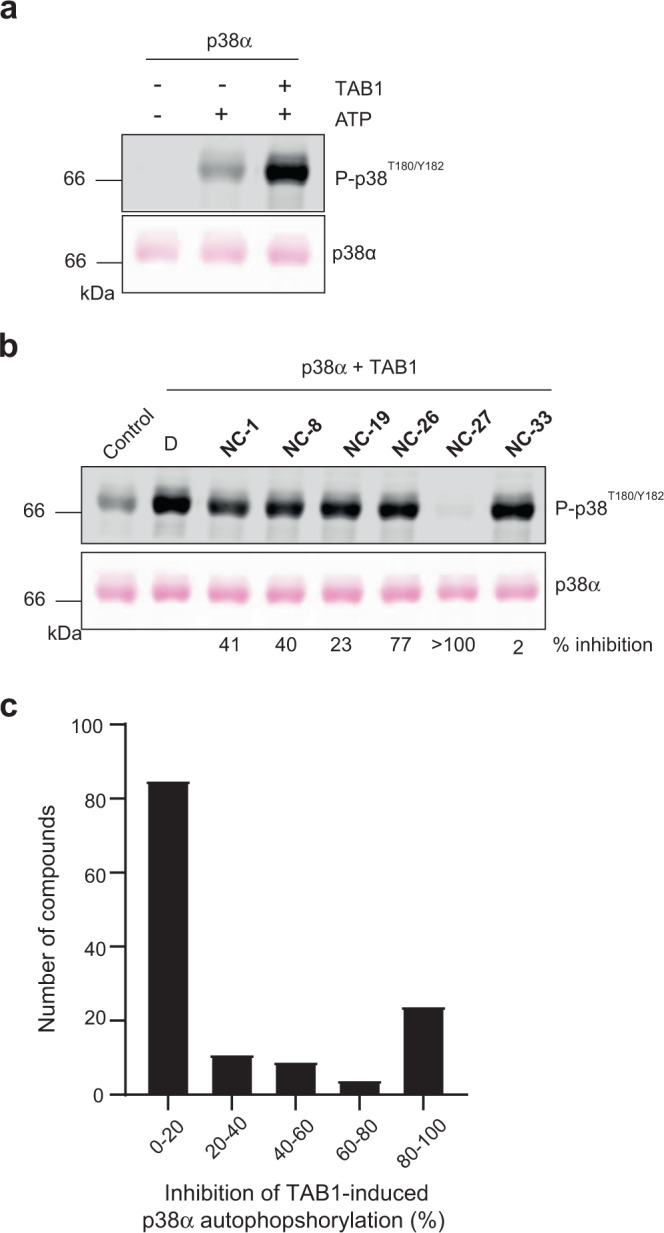
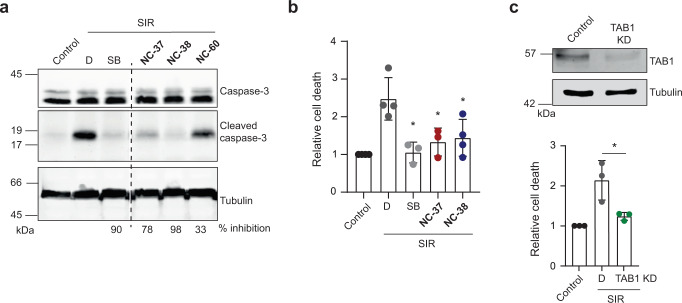
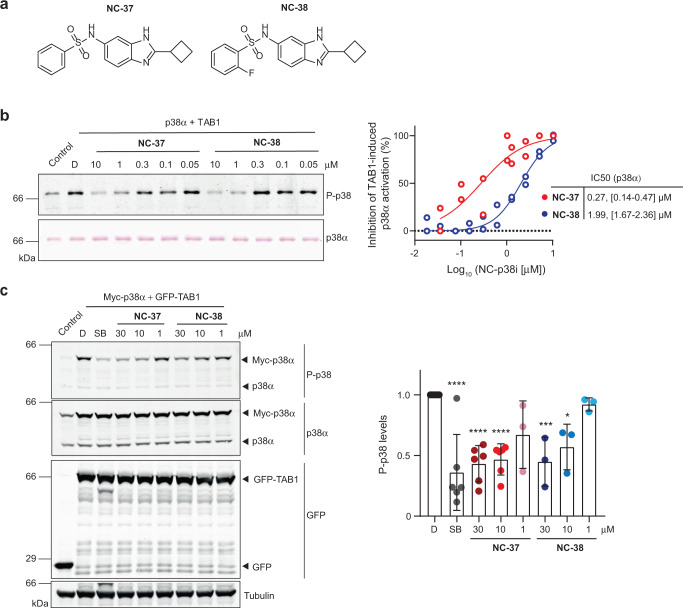
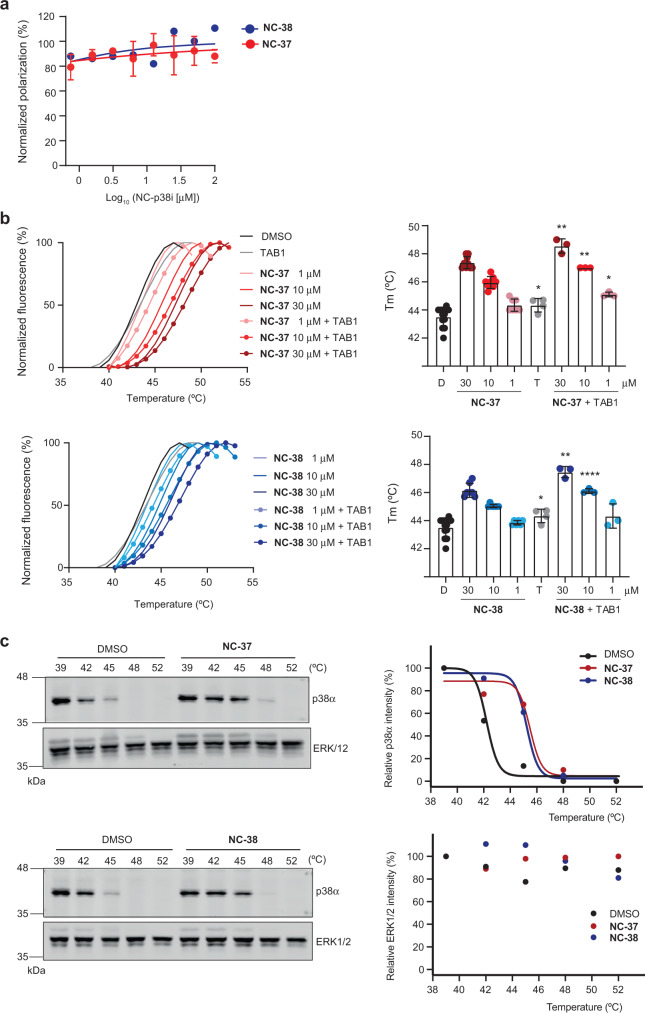
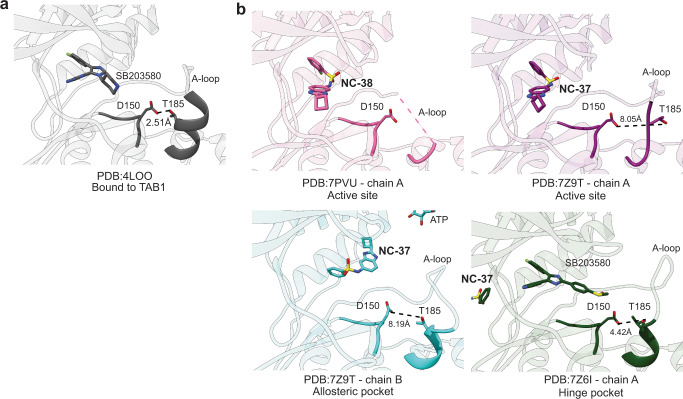
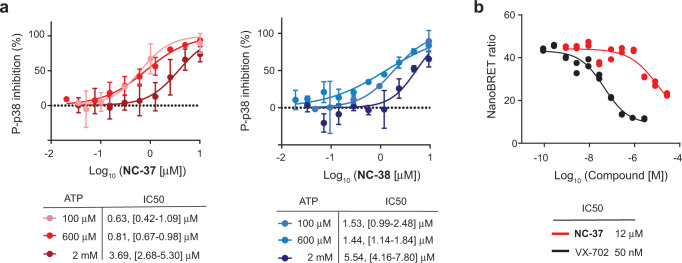
p38α is a versatile protein kinase that can control numerous processes and plays important roles in the cellular responses to stress. Dysregulation of p38α signaling has been linked to several diseases including inflammation, immune disorders and cancer, suggesting that targeting p38α could be therapeutically beneficial. Over the last two decades, numerous p38α inhibitors have been developed, which showed promising effects in pre-clinical studies but results from clinical trials have been disappointing, fueling the interest in the generation of alternative mechanisms of p38α modulation. Here, we report the in silico identification of compounds that we refer to as non-canonical p38α inhibitors (NC-p38i). By combining biochemical and structural analyses, we show that NC-p38i efficiently inhibit p38α autophosphorylation but weakly affect the activity of the canonical pathway. Our results demonstrate how the structural plasticity of p38α can be leveraged to develop therapeutic opportunities targeting a subset of the functions regulated by this pathway.
© 2023. The Author(s).
Conflict of interest statement
IRB Barcelona, UB, ICREA, BSC-CNS, and Nostrum Biodiscovery have filed the patent application WO2020120576 - P38Α AUTOPHOSPHORYLATION INHIBITORS. L.G., L.D., A.I., R.S., M.O., and A.R.N. are named inventors on this application, but the patent was abandoned. The rest of the authors declare no competing interests.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
