

H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function
- PMID: 37308596
- PMCID: PMC10352135
- DOI: 10.1038/s41594-023-01016-5
H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function
Abstract
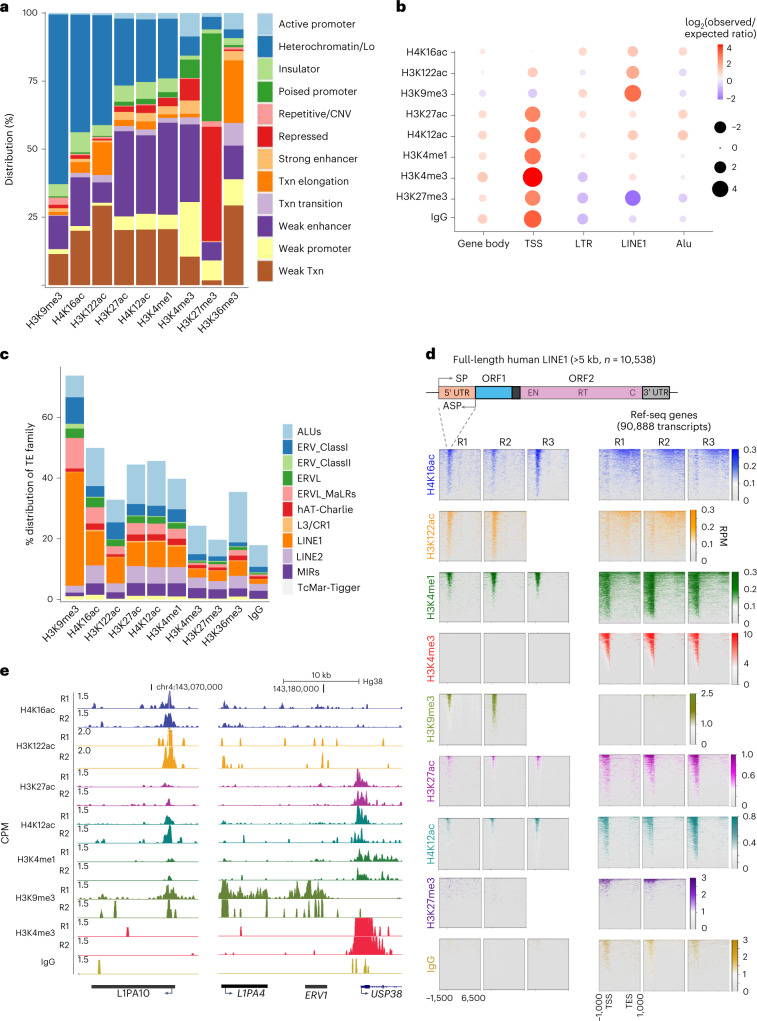
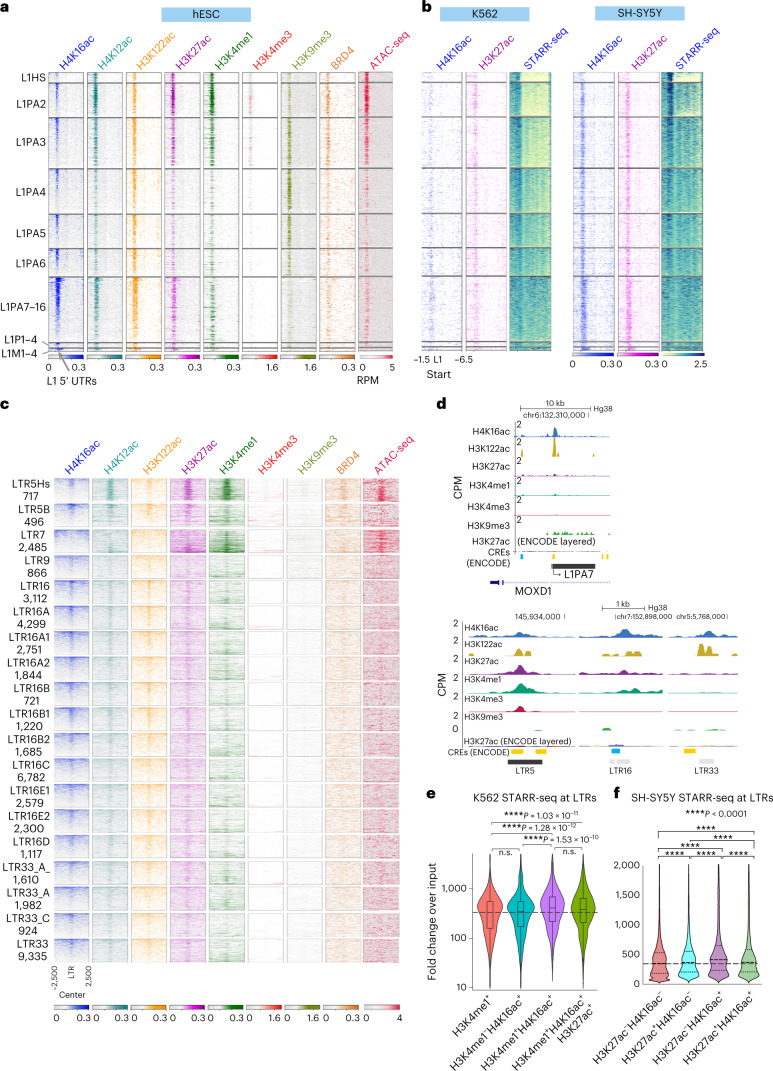
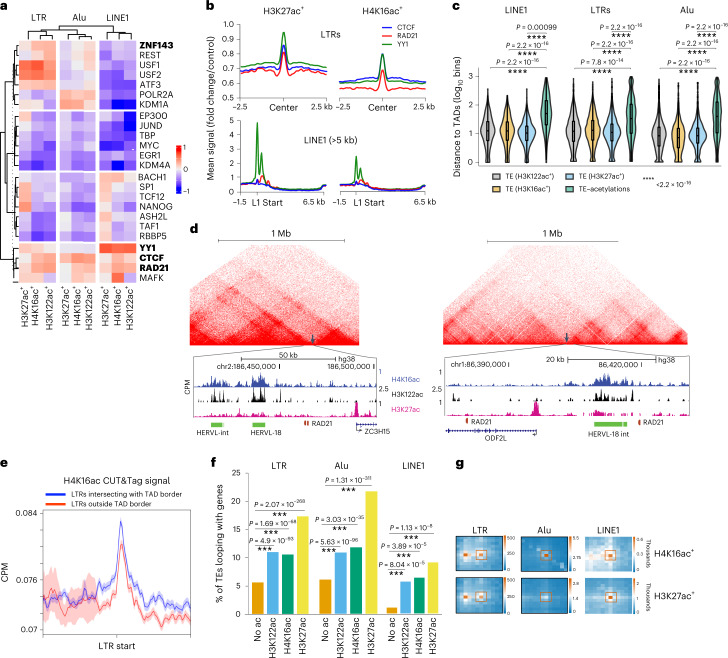
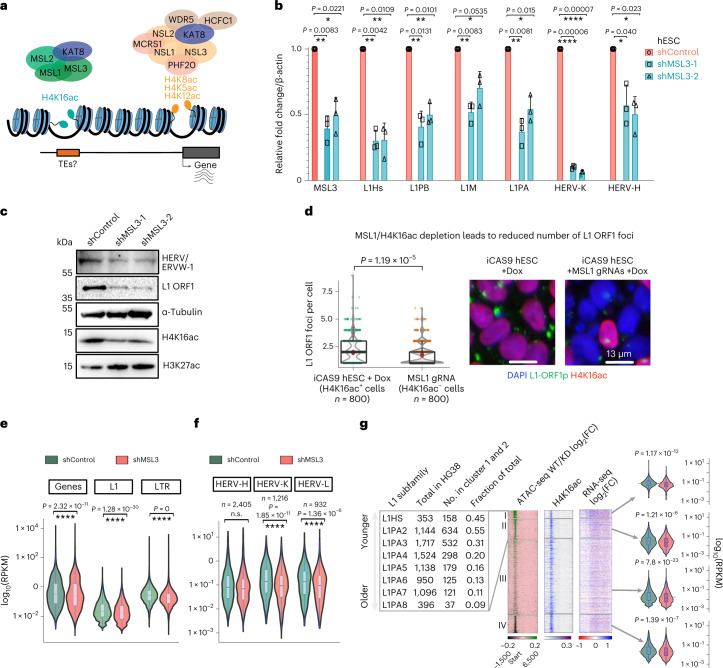
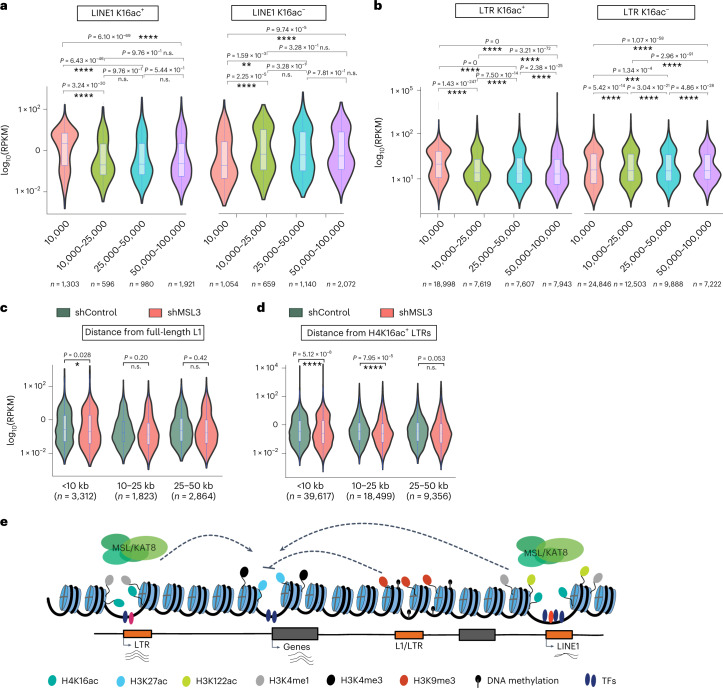
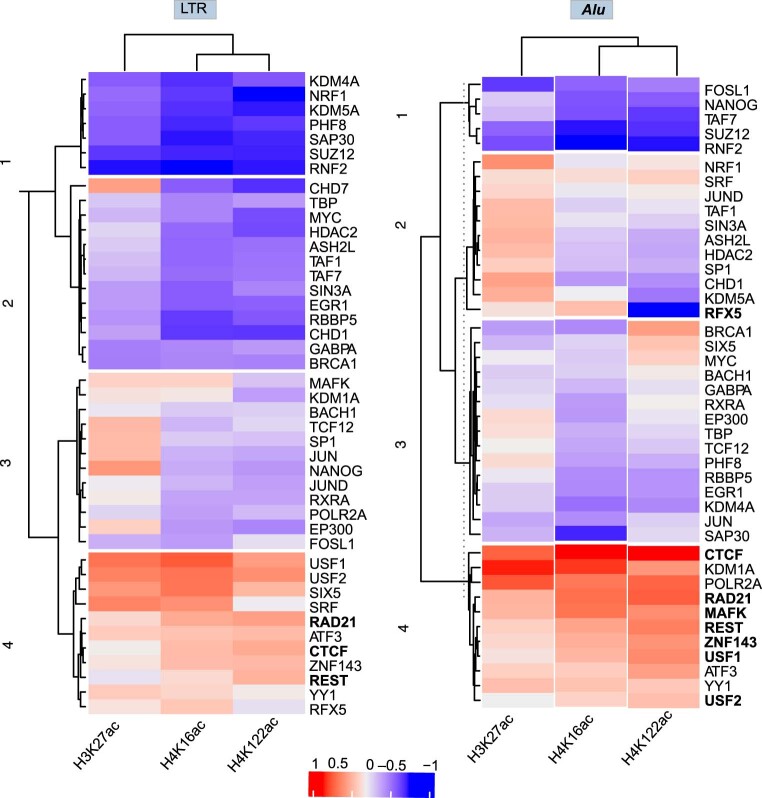
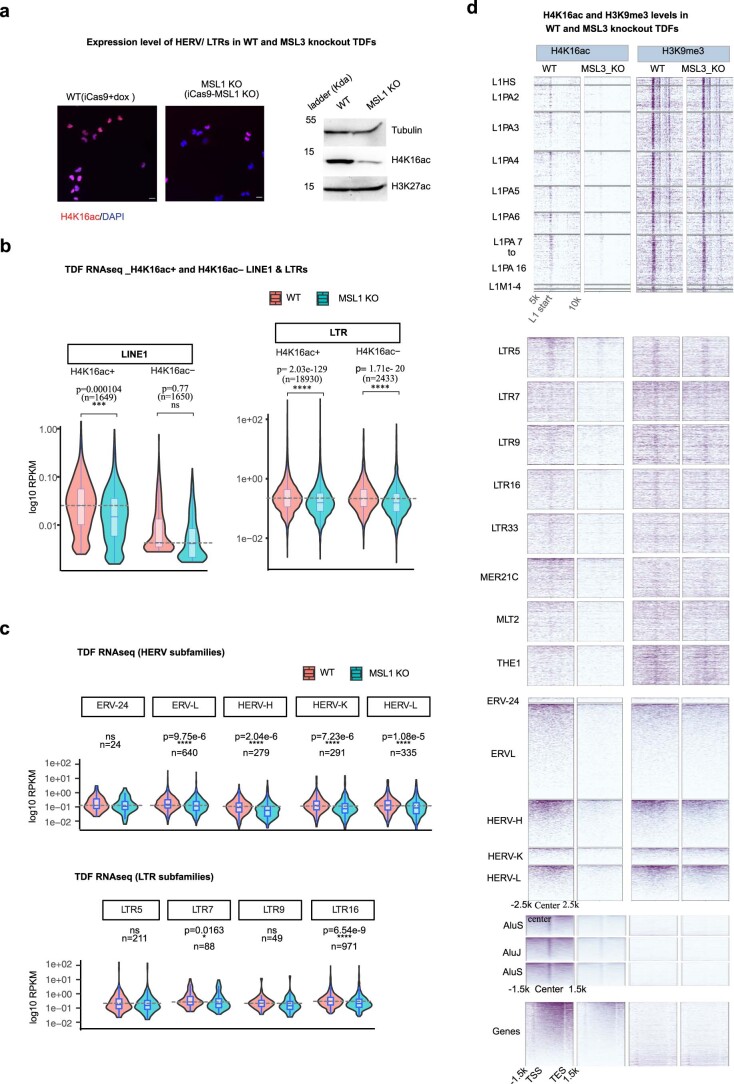
Mammalian genomes harbor abundant transposable elements (TEs) and their remnants, with numerous epigenetic repression mechanisms enacted to silence TE transcription. However, TEs are upregulated during early development, neuronal lineage, and cancers, although the epigenetic factors contributing to the transcription of TEs have yet to be fully elucidated. Here, we demonstrate that the male-specific lethal (MSL)-complex-mediated histone H4 acetylation at lysine 16 (H4K16ac) is enriched at TEs in human embryonic stem cells (hESCs) and cancer cells. This in turn activates transcription of subsets of full-length long interspersed nuclear elements (LINE1s, L1s) and endogenous retrovirus (ERV) long terminal repeats (LTRs). Furthermore, we show that the H4K16ac-marked L1 and LTR subfamilies display enhancer-like functions and are enriched in genomic locations with chromatin features associated with active enhancers. Importantly, such regions often reside at boundaries of topologically associated domains and loop with genes. CRISPR-based epigenetic perturbation and genetic deletion of L1s reveal that H4K16ac-marked L1s and LTRs regulate the expression of genes in cis. Overall, TEs enriched with H4K16ac contribute to the cis-regulatory landscape at specific genomic locations by maintaining an active chromatin landscape at TEs.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
