

Expanding genotype-phenotype correlations in FOXG1 syndrome: results from a patient registry
- PMID: 37308910
- PMCID: PMC10262363
- DOI: 10.1186/s13023-023-02745-y
Expanding genotype-phenotype correlations in FOXG1 syndrome: results from a patient registry
Abstract
Background: We refine the clinical spectrum of FOXG1 syndrome and expand genotype-phenotype correlations through evaluation of 122 individuals enrolled in an international patient registry.
Methods: The FOXG1 syndrome online patient registry allows for remote collection of caregiver-reported outcomes. Inclusion required documentation of a (likely) pathogenic variant in FOXG1. Caregivers were administered a questionnaire to evaluate clinical severity of core features of FOXG1 syndrome. Genotype-phenotype correlations were determined using nonparametric analyses.
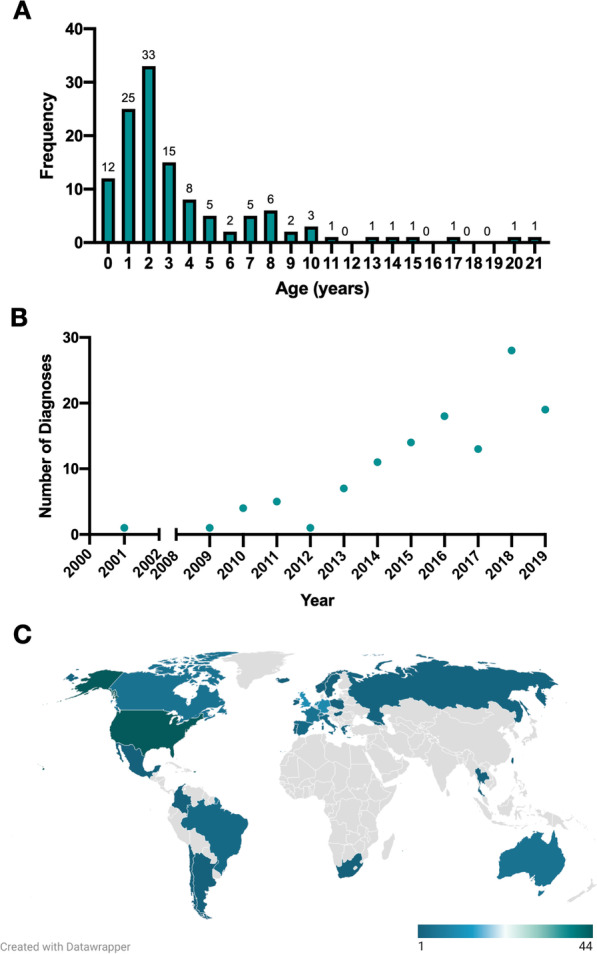
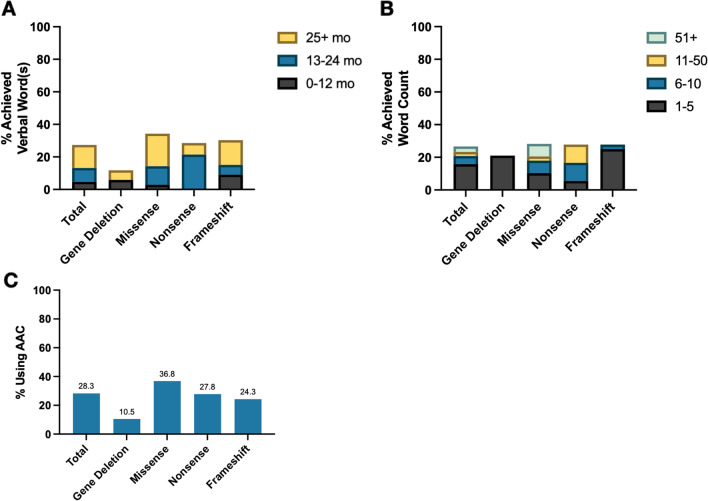
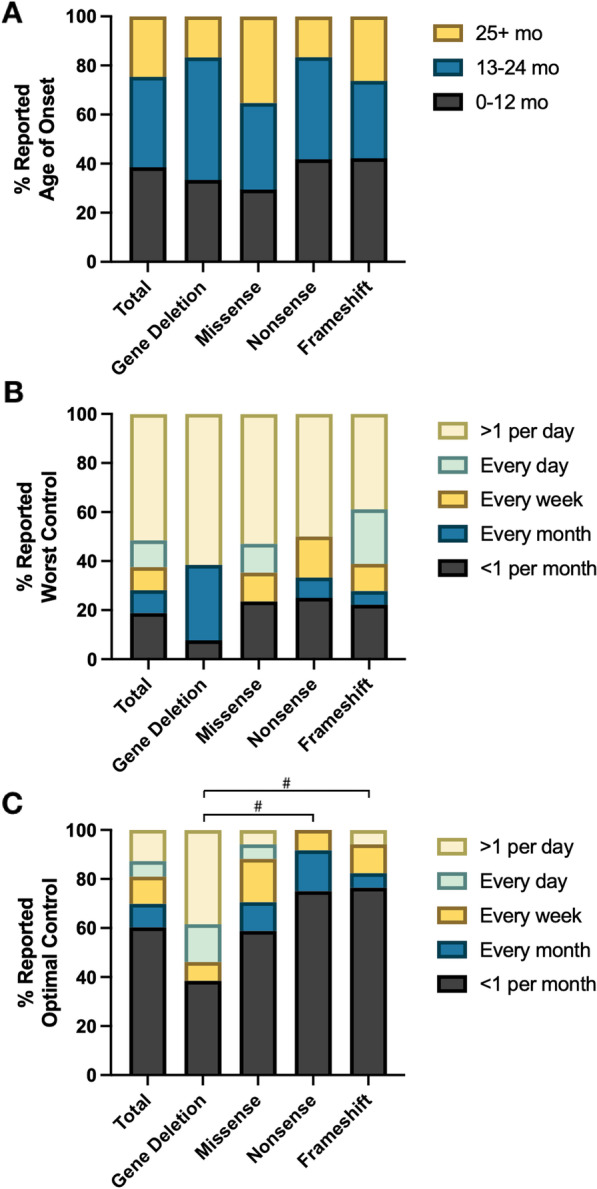
Results: We studied 122 registry participants with FOXG1 syndrome, aged < 12 months to 24 years. Caregivers described delayed or absent developmental milestone attainment, seizures (61%), and movement disorders (58%). Participants harbouring a missense variant had a milder phenotype. Compared to individuals with gene deletions (0%) or nonsense variants (20%), missense variants were associated with more frequent attainment of sitting (73%). Further, individuals with missense variants (41%) achieved independent walking more frequently than those with gene deletions (0%) or frameshift variants (6%). Presence of epilepsy also varied by genotype and was significantly more common in those with gene deletions (81%) compared to missense variants (47%). Individuals with gene deletions were more likely to have higher seizure burden than other genotypes with 53% reporting daily seizures, even at best control. We also observed that truncations preserving the forkhead DNA binding domain were associated with better developmental outcomes.
Conclusion: We refine the phenotypic spectrum of neurodevelopmental features associated with FOXG1 syndrome. We strengthen genotype-driven outcomes, where missense variants are associated with a milder clinical course.
Keywords: Epilepsy; Genotype–phenotype association; Movement Disorder; Patient registry; Rare neurological diseases.
© 2023. The Author(s).
Conflict of interest statement
There are no competing of interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
