

Embryonic vitamin D deficiency programs hematopoietic stem cells to induce type 2 diabetes
- PMID: 37311757
- PMCID: PMC10264405
- DOI: 10.1038/s41467-023-38849-z
Embryonic vitamin D deficiency programs hematopoietic stem cells to induce type 2 diabetes
Abstract
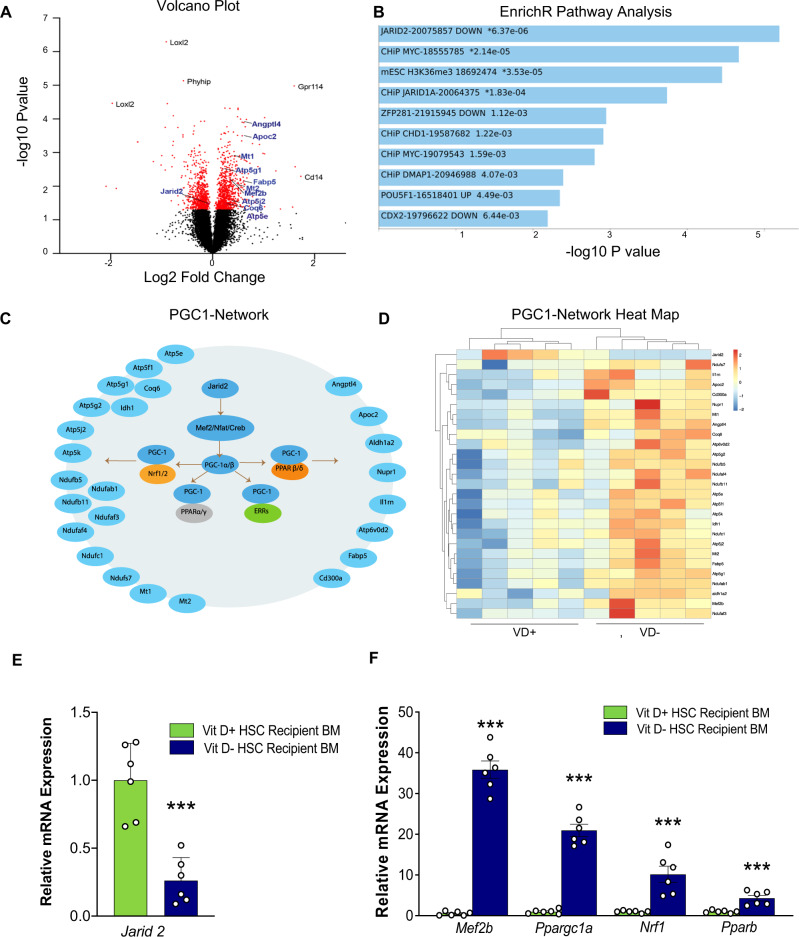
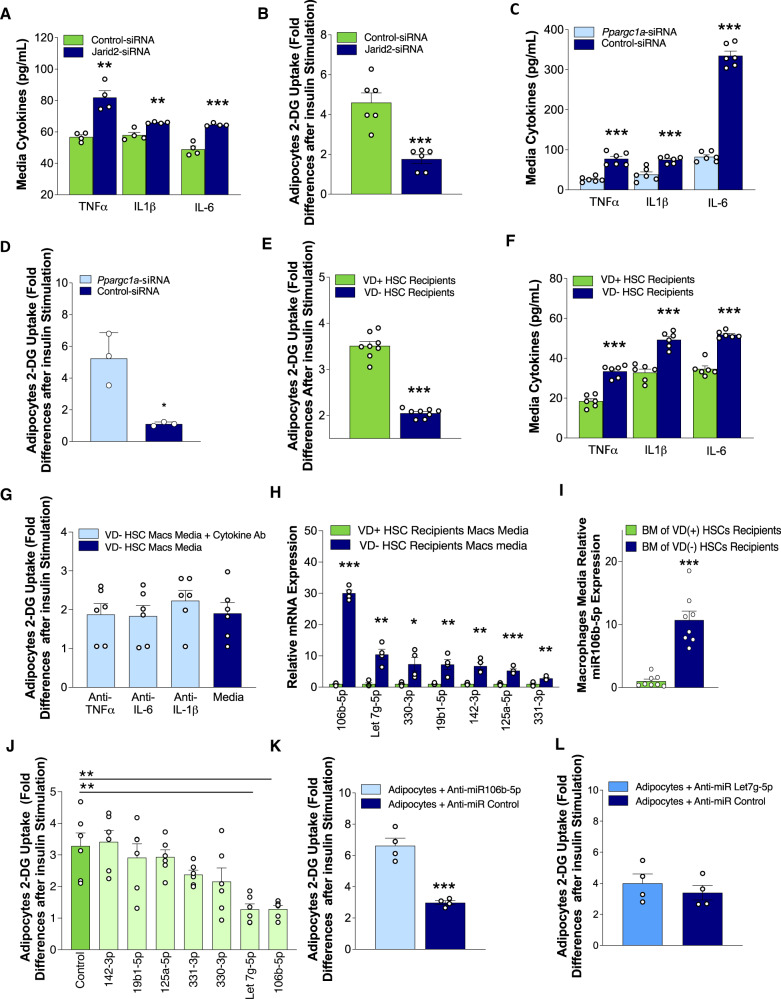
Environmental factors may alter the fetal genome to cause metabolic diseases. It is unknown whether embryonic immune cell programming impacts the risk of type 2 diabetes in later life. We demonstrate that transplantation of fetal hematopoietic stem cells (HSCs) made vitamin D deficient in utero induce diabetes in vitamin D-sufficient mice. Vitamin D deficiency epigenetically suppresses Jarid2 expression and activates the Mef2/PGC1a pathway in HSCs, which persists in recipient bone marrow, resulting in adipose macrophage infiltration. These macrophages secrete miR106-5p, which promotes adipose insulin resistance by repressing PIK3 catalytic and regulatory subunits and down-regulating AKT signaling. Vitamin D-deficient monocytes from human cord blood have comparable Jarid2/Mef2/PGC1a expression changes and secrete miR-106b-5p, causing adipocyte insulin resistance. These findings suggest that vitamin D deficiency during development has epigenetic consequences impacting the systemic metabolic milieu.
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and ItsBurden in the United States, 2014. (Department of Health and Human Services, Atlanta, GA, 2014).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
