

A pangenome reference of 36 Chinese populations
- PMID: 37316654
- PMCID: PMC10322713
- DOI: 10.1038/s41586-023-06173-7
A pangenome reference of 36 Chinese populations
Abstract
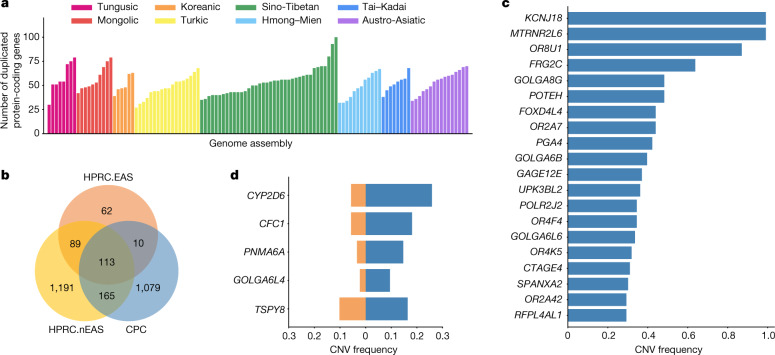
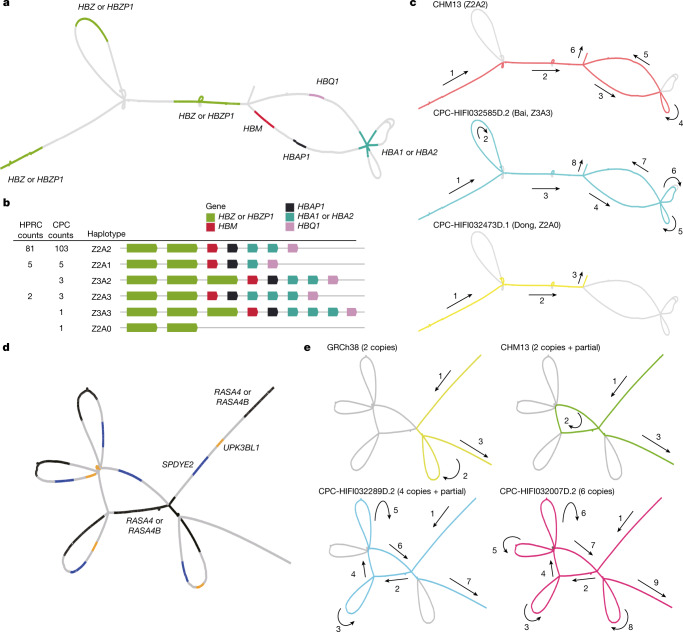
Human genomics is witnessing an ongoing paradigm shift from a single reference sequence to a pangenome form, but populations of Asian ancestry are underrepresented. Here we present data from the first phase of the Chinese Pangenome Consortium, including a collection of 116 high-quality and haplotype-phased de novo assemblies based on 58 core samples representing 36 minority Chinese ethnic groups. With an average 30.65× high-fidelity long-read sequence coverage, an average contiguity N50 of more than 35.63 megabases and an average total size of 3.01 gigabases, the CPC core assemblies add 189 million base pairs of euchromatic polymorphic sequences and 1,367 protein-coding gene duplications to GRCh38. We identified 15.9 million small variants and 78,072 structural variants, of which 5.9 million small variants and 34,223 structural variants were not reported in a recently released pangenome reference1. The Chinese Pangenome Consortium data demonstrate a remarkable increase in the discovery of novel and missing sequences when individuals are included from underrepresented minority ethnic groups. The missing reference sequences were enriched with archaic-derived alleles and genes that confer essential functions related to keratinization, response to ultraviolet radiation, DNA repair, immunological responses and lifespan, implying great potential for shedding new light on human evolution and recovering missing heritability in complex disease mapping.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












Comment in
-
Public perceptions of international genetic information sharing for biomedical research in China: a case study of the social media debate on the article "A Pangenome Reference of 36 Chinese Populations" published in Nature.Hum Genomics. 2024 Aug 7;18(1):86. doi: 10.1186/s40246-024-00650-4. Hum Genomics. 2024. PMID: 39113147 Free PMC article.
References
-
- Liao, W.-W. et al. A draft human pangenome reference. Preprint at 10.1101/2022.07.09.499321 (2022).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
