

CNK2 promotes cancer cell motility by mediating ARF6 activation downstream of AXL signalling
- PMID: 37322019
- PMCID: PMC10272126
- DOI: 10.1038/s41467-023-39281-z
CNK2 promotes cancer cell motility by mediating ARF6 activation downstream of AXL signalling
Abstract
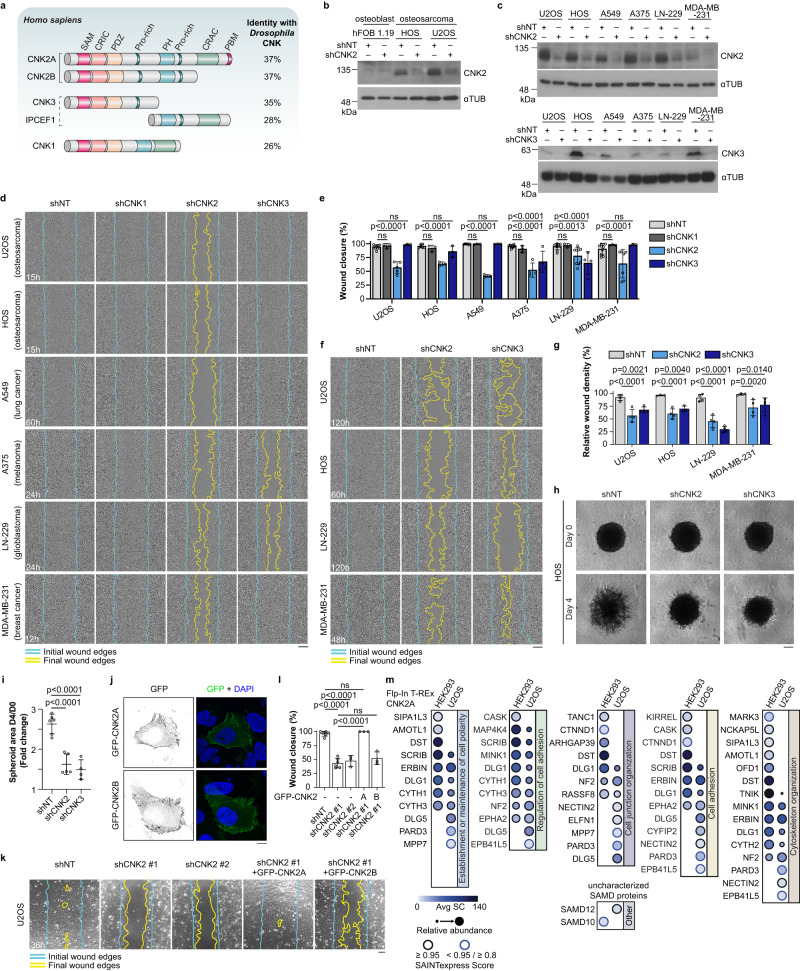
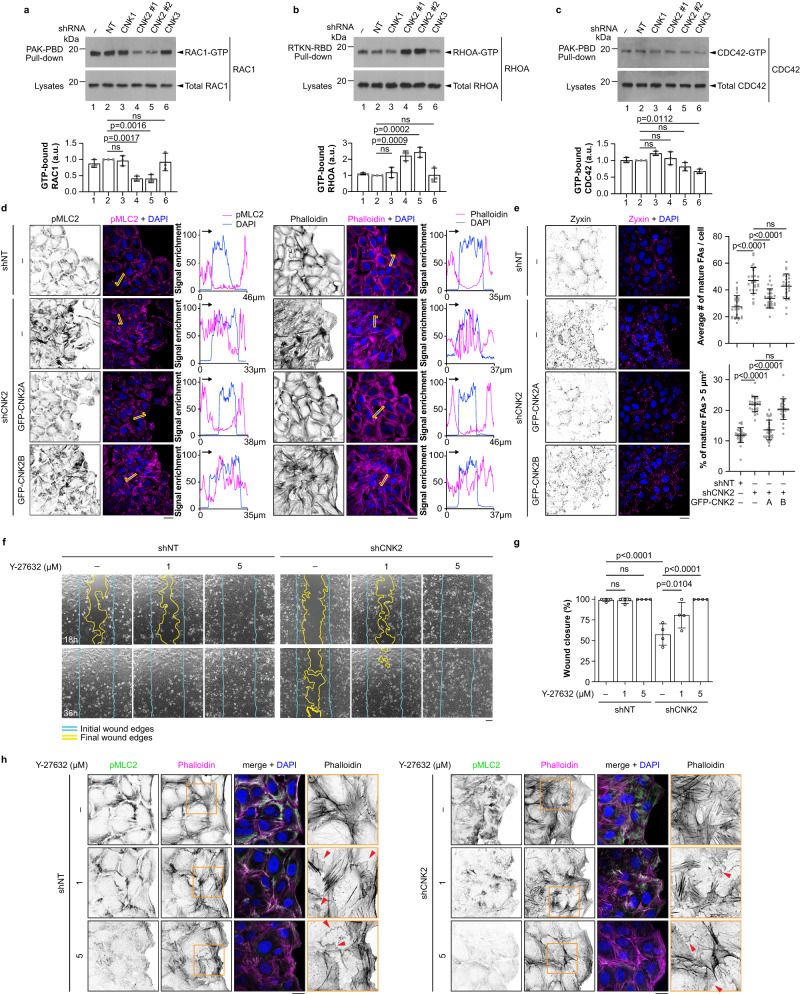
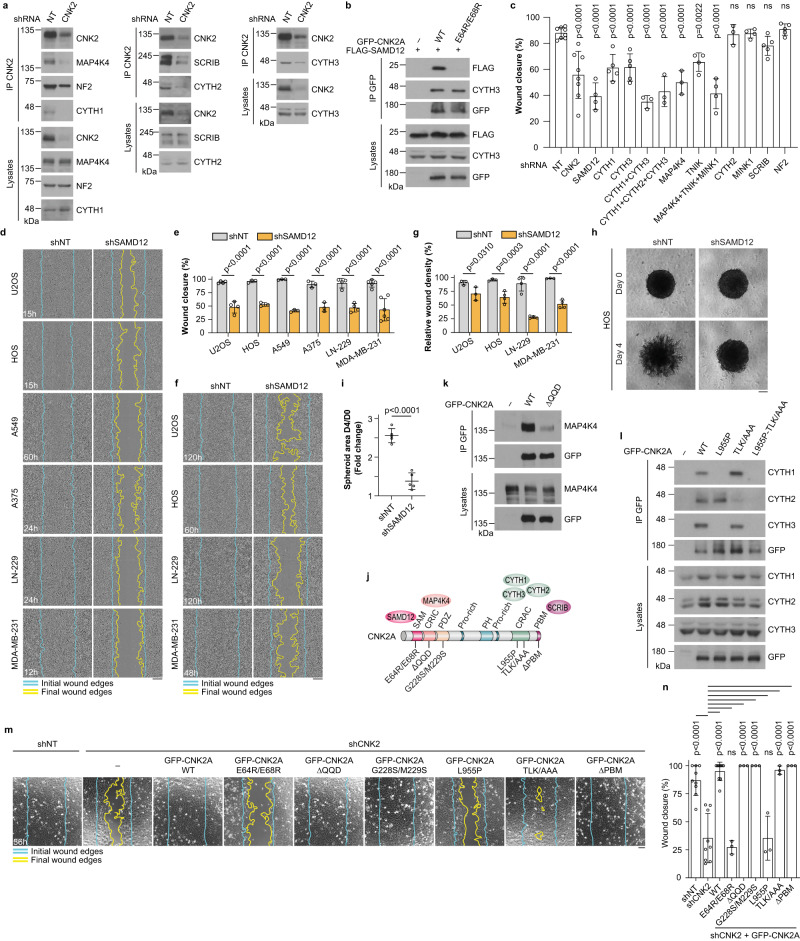
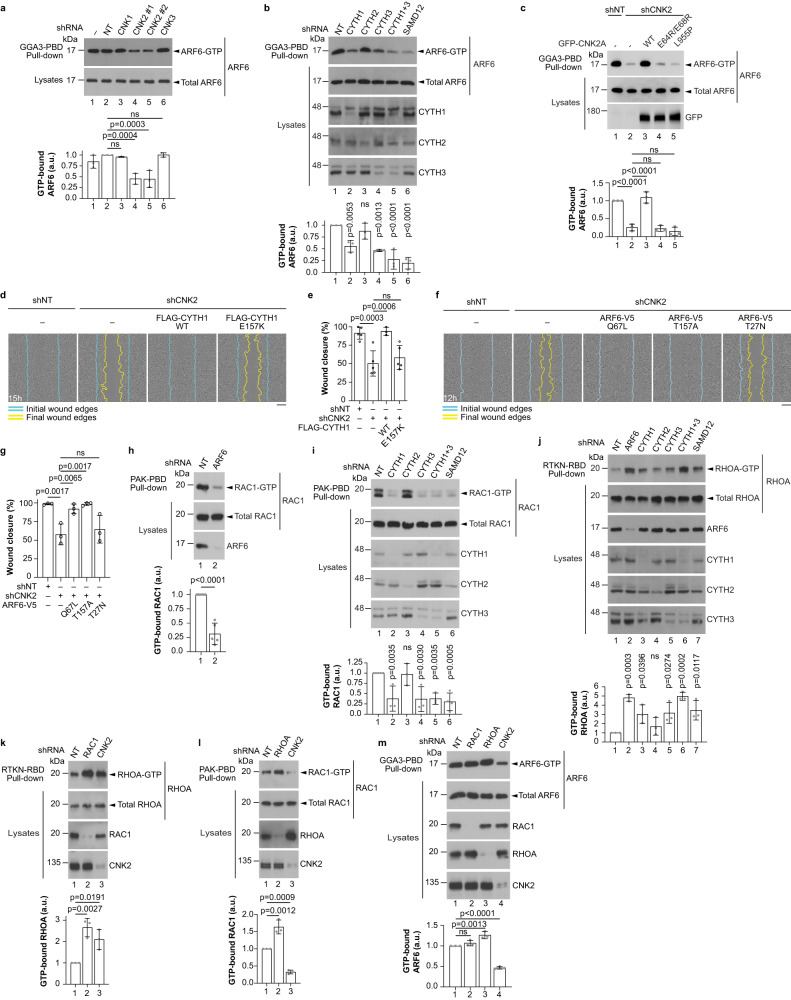
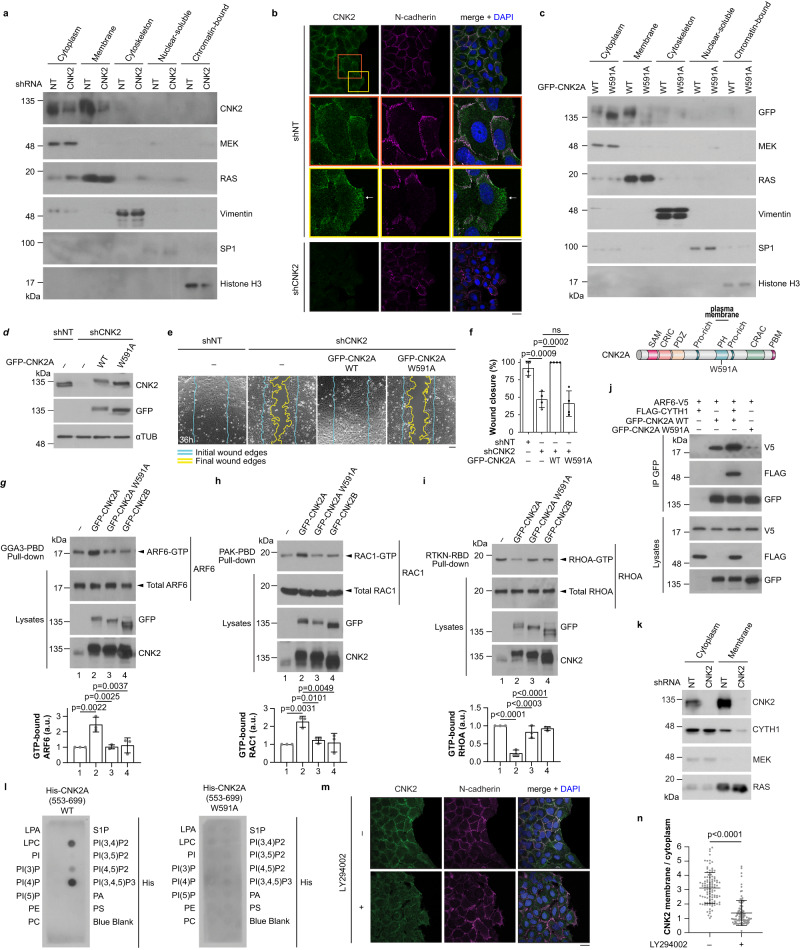
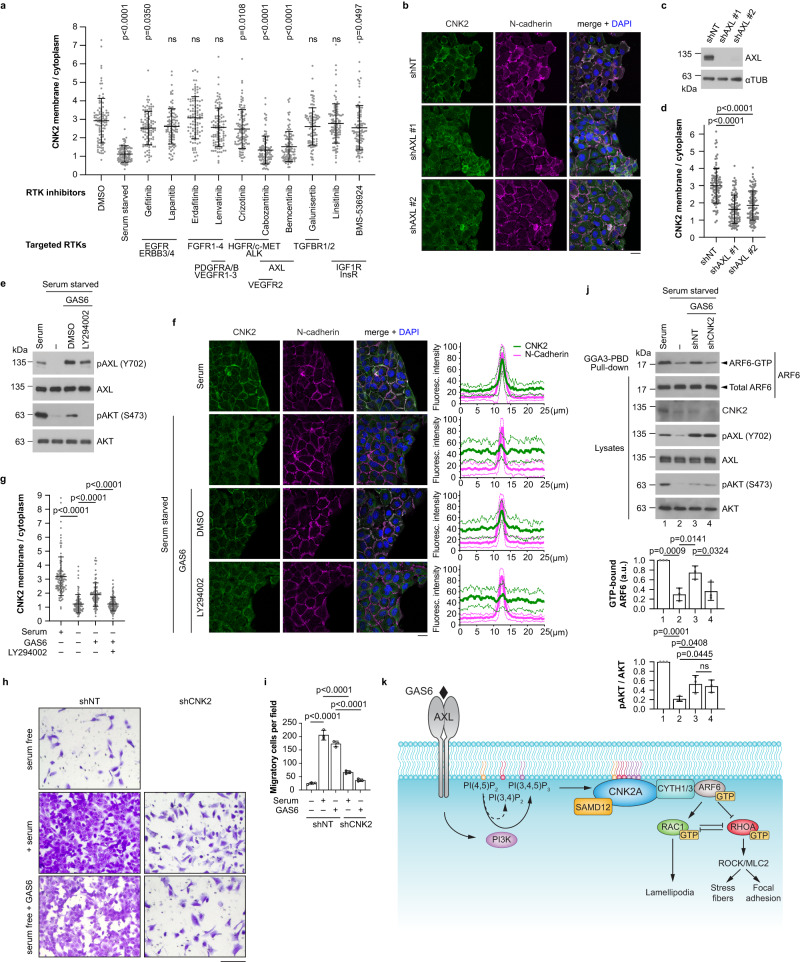
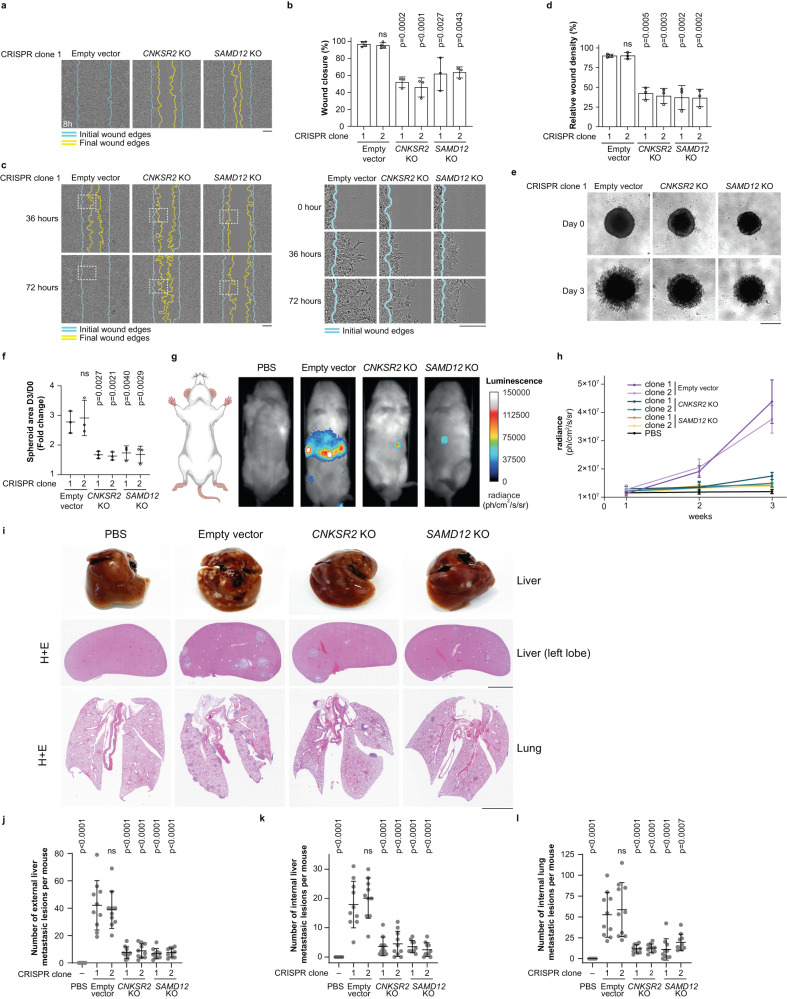
Cell motility is a critical feature of invasive tumour cells that is governed by complex signal transduction events. Particularly, the underlying mechanisms that bridge extracellular stimuli to the molecular machinery driving motility remain partially understood. Here, we show that the scaffold protein CNK2 promotes cancer cell migration by coupling the pro-metastatic receptor tyrosine kinase AXL to downstream activation of ARF6 GTPase. Mechanistically, AXL signalling induces PI3K-dependent recruitment of CNK2 to the plasma membrane. In turn, CNK2 stimulates ARF6 by associating with cytohesin ARF GEFs and with a novel adaptor protein called SAMD12. ARF6-GTP then controls motile forces by coordinating the respective activation and inhibition of RAC1 and RHOA GTPases. Significantly, genetic ablation of CNK2 or SAMD12 reduces metastasis in a mouse xenograft model. Together, this work identifies CNK2 and its partner SAMD12 as key components of a novel pro-motility pathway in cancer cells, which could be targeted in metastasis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
ARF6 activated by the LHCG receptor through the cytohesin family of guanine nucleotide exchange factors mediates the receptor internalization and signaling.J Biol Chem. 2012 Jun 8;287(24):20443-55. doi: 10.1074/jbc.M112.362087. Epub 2012 Apr 20. J Biol Chem. 2012. PMID: 22523074 Free PMC article.
-
ARF6-Rac1 signaling-mediated neurite outgrowth is potentiated by the neuronal adaptor FE65 through orchestrating ARF6 and ELMO1.FASEB J. 2020 Dec;34(12):16397-16413. doi: 10.1096/fj.202001703R. Epub 2020 Oct 13. FASEB J. 2020. PMID: 33047393
-
ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway.Cancer Res. 2009 Feb 1;69(3):794-801. doi: 10.1158/0008-5472.CAN-08-2110. Epub 2009 Jan 20. Cancer Res. 2009. PMID: 19155310 Free PMC article.
-
Centaurin-alpha1 and KIF13B kinesin motor protein interaction in ARF6 signalling.Biochem Soc Trans. 2005 Dec;33(Pt 6):1279-81. doi: 10.1042/BST0331279. Biochem Soc Trans. 2005. PMID: 16246098 Review.
-
Potential regulation of ADP-ribosylation factor 6 signalling by phosphatidylinositol 3,4,5-trisphosphate.Biochem Soc Trans. 1999 Aug;27(4):683-9. doi: 10.1042/bst0270683. Biochem Soc Trans. 1999. PMID: 10917667 Review.
Cited by
-
The CNK-HYP scaffolding complex promotes RAF activation by enhancing KSR-MEK interaction.Nat Struct Mol Biol. 2024 Jul;31(7):1028-1038. doi: 10.1038/s41594-024-01233-6. Epub 2024 Feb 22. Nat Struct Mol Biol. 2024. PMID: 38388830 Free PMC article.
-
Mutant PP2A Induces IGFBP2 Secretion to Promote Development of High-Grade Uterine Cancer.Cancer Res. 2025 Feb 1;85(3):442-461. doi: 10.1158/0008-5472.CAN-24-1263. Cancer Res. 2025. PMID: 39531506 Free PMC article.
-
Exploring the Potential of Oleanolic Acid Dimers-Cytostatic and Antioxidant Activities, Molecular Docking, and ADMETox Profile.Molecules. 2024 Jul 31;29(15):3623. doi: 10.3390/molecules29153623. Molecules. 2024. PMID: 39125028 Free PMC article.
-
Role and therapeutic potential of the NEDD4 family in breast cancer.Front Pharmacol. 2025 Jun 4;16:1587675. doi: 10.3389/fphar.2025.1587675. eCollection 2025. Front Pharmacol. 2025. PMID: 40535763 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
