

A complete telomere-to-telomere assembly of the maize genome
- PMID: 37322109
- PMCID: PMC10335936
- DOI: 10.1038/s41588-023-01419-6
A complete telomere-to-telomere assembly of the maize genome
Abstract
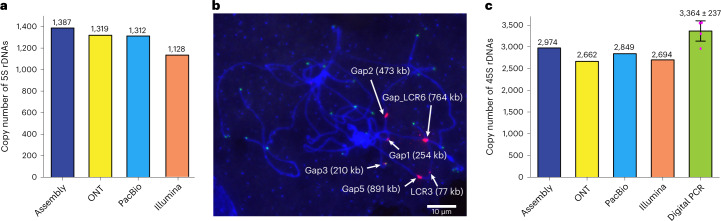
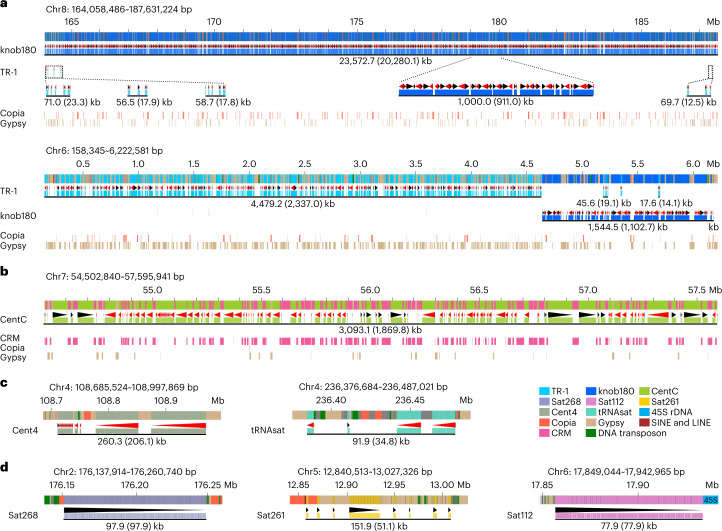
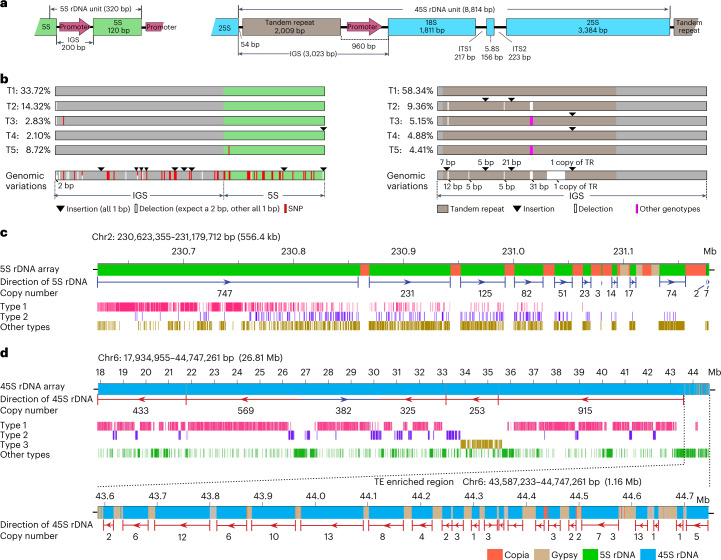
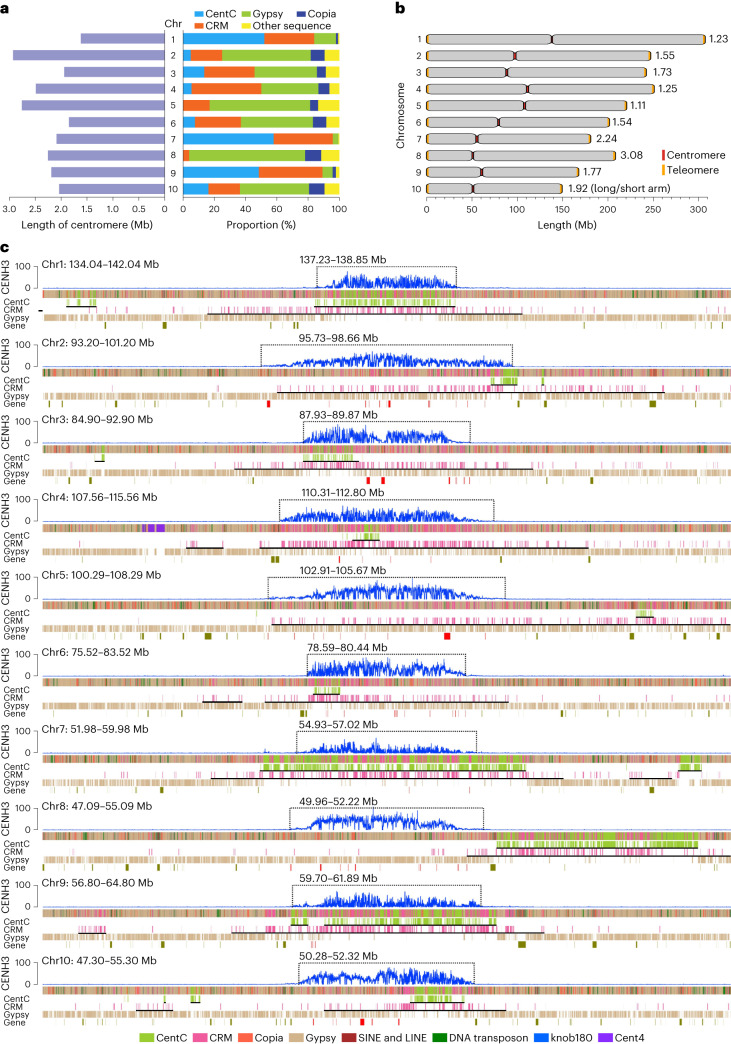
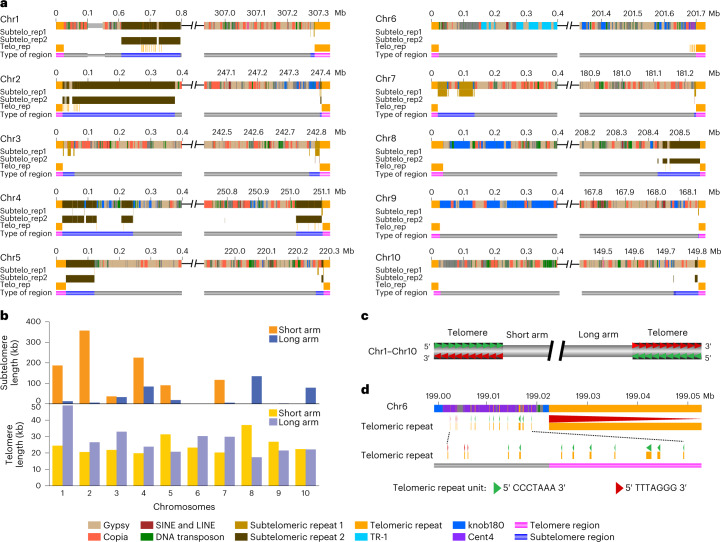
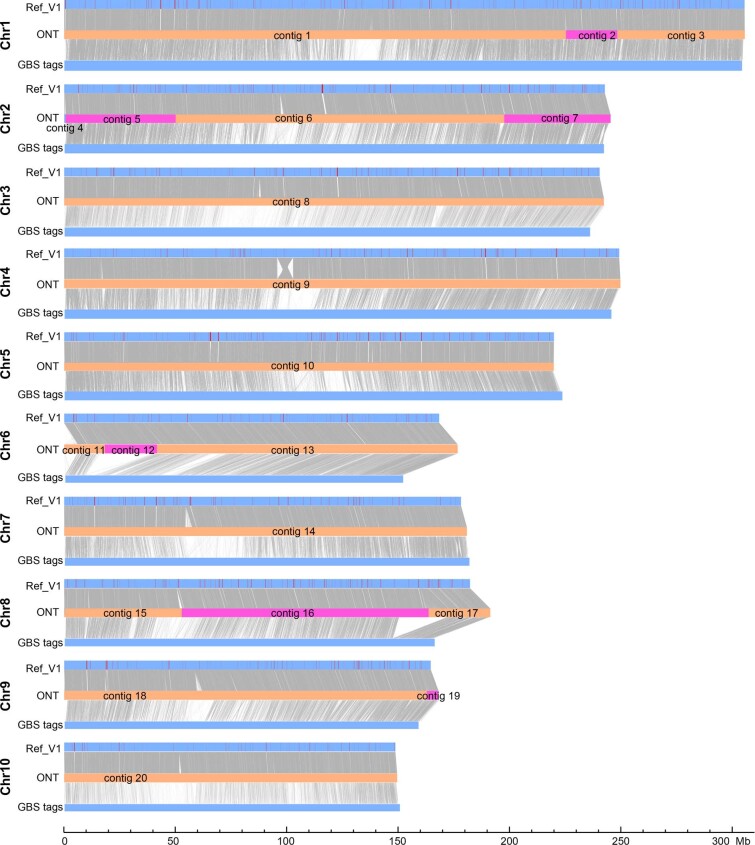
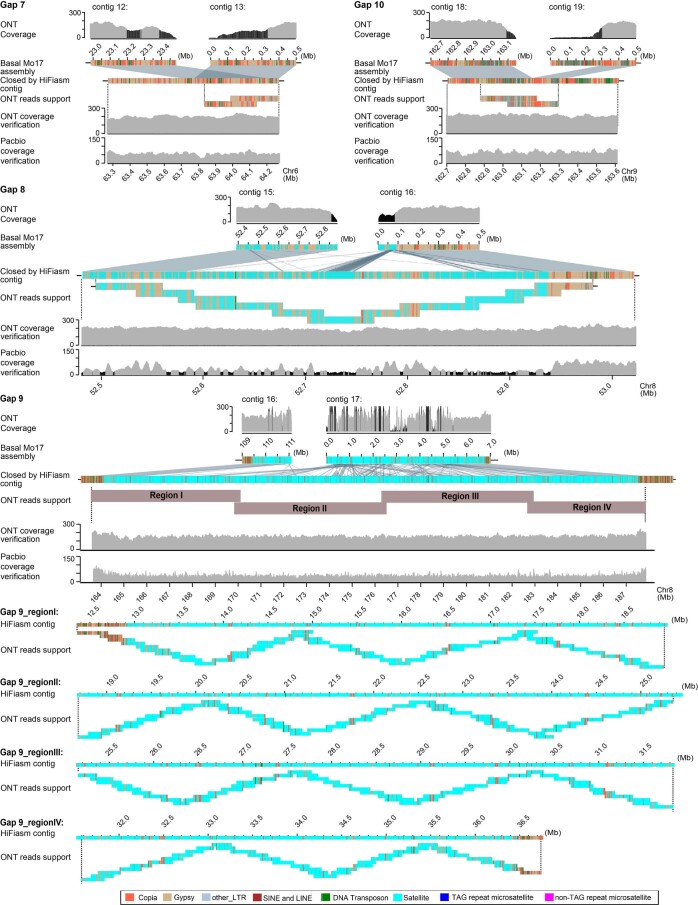
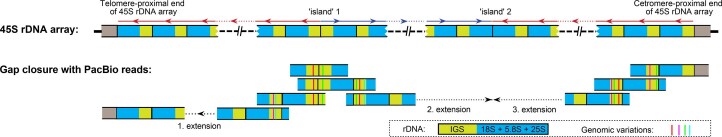
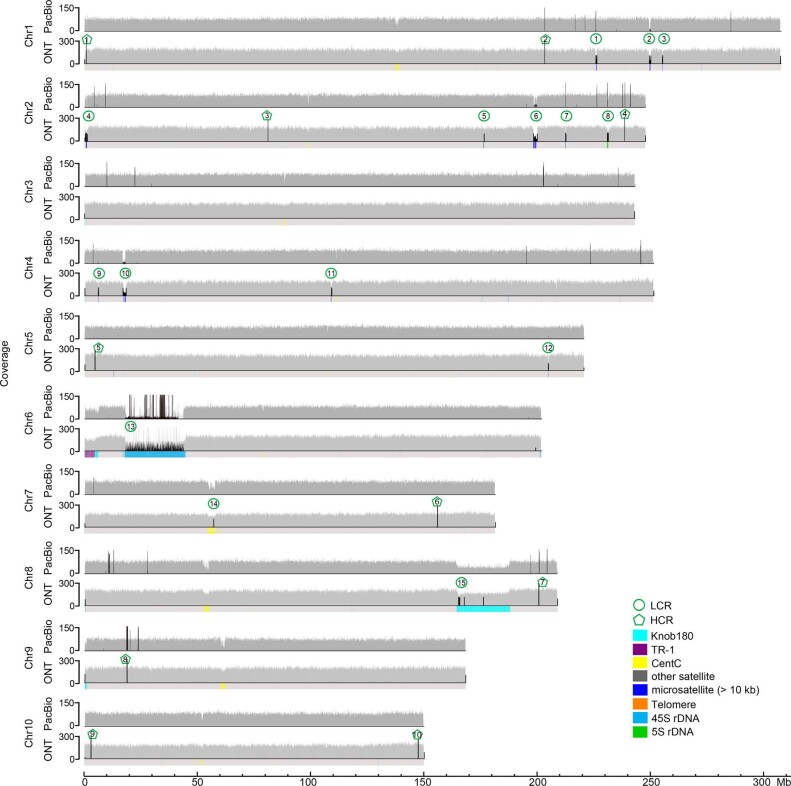
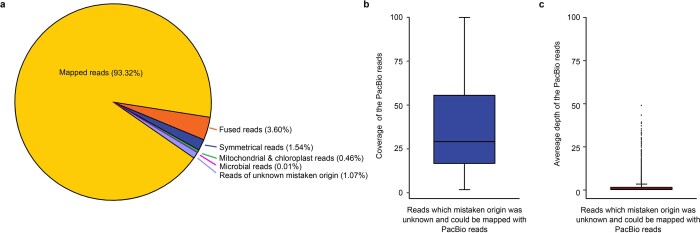
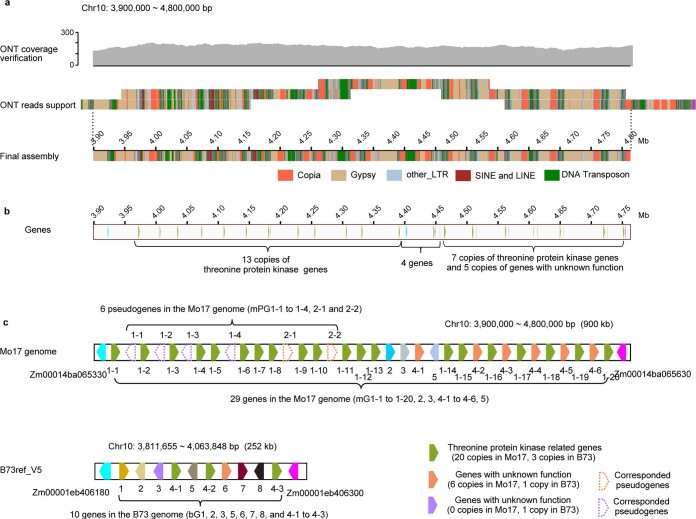
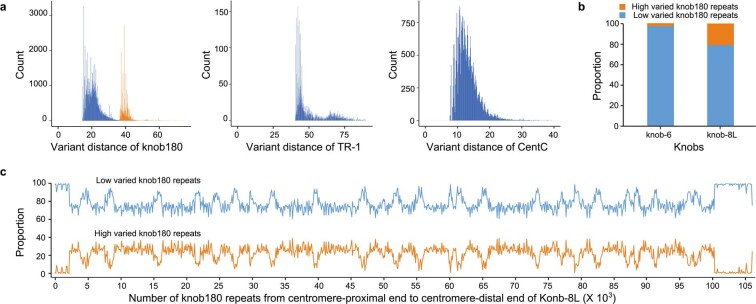
A complete telomere-to-telomere (T2T) finished genome has been the long pursuit of genomic research. Through generating deep coverage ultralong Oxford Nanopore Technology (ONT) and PacBio HiFi reads, we report here a complete genome assembly of maize with each chromosome entirely traversed in a single contig. The 2,178.6 Mb T2T Mo17 genome with a base accuracy of over 99.99% unveiled the structural features of all repetitive regions of the genome. There were several super-long simple-sequence-repeat arrays having consecutive thymine-adenine-guanine (TAG) tri-nucleotide repeats up to 235 kb. The assembly of the entire nucleolar organizer region of the 26.8 Mb array with 2,974 45S rDNA copies revealed the enormously complex patterns of rDNA duplications and transposon insertions. Additionally, complete assemblies of all ten centromeres enabled us to precisely dissect the repeat compositions of both CentC-rich and CentC-poor centromeres. The complete Mo17 genome represents a major step forward in understanding the complexity of the highly recalcitrant repetitive regions of higher plant genomes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature408, 796 (2000). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
