

Direct enzymatic sequencing of 5-methylcytosine at single-base resolution
- PMID: 37322153
- PMCID: PMC10763687
- DOI: 10.1038/s41589-023-01318-1
Direct enzymatic sequencing of 5-methylcytosine at single-base resolution
Abstract
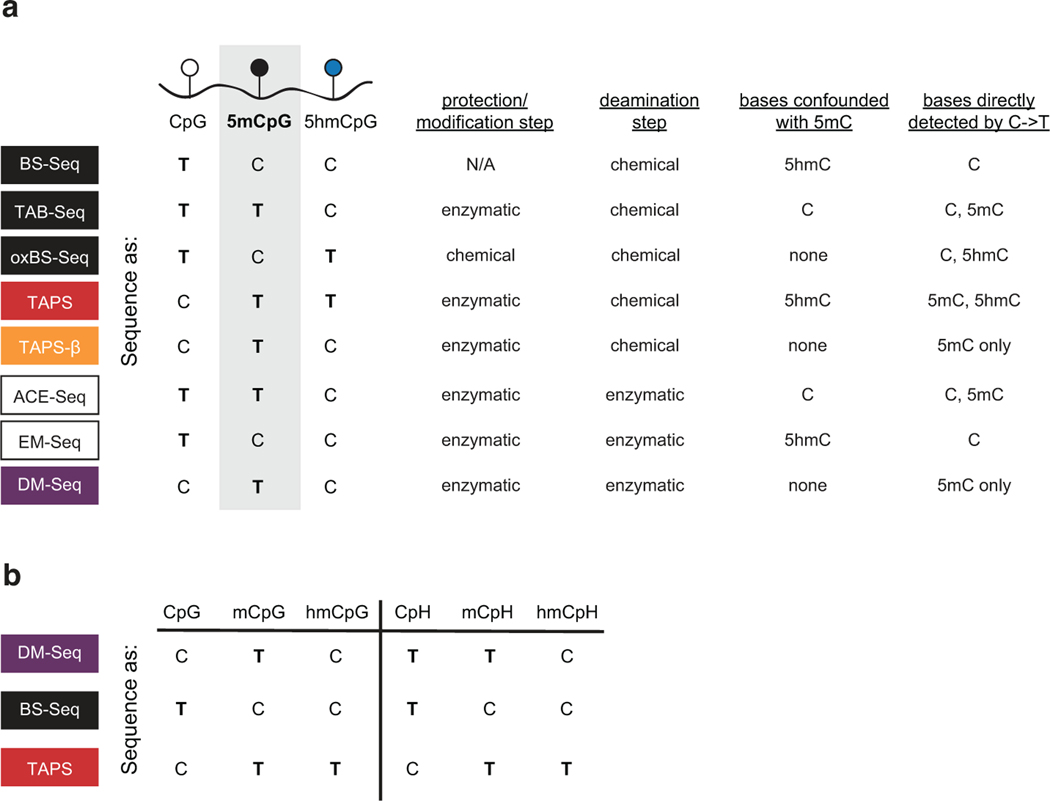
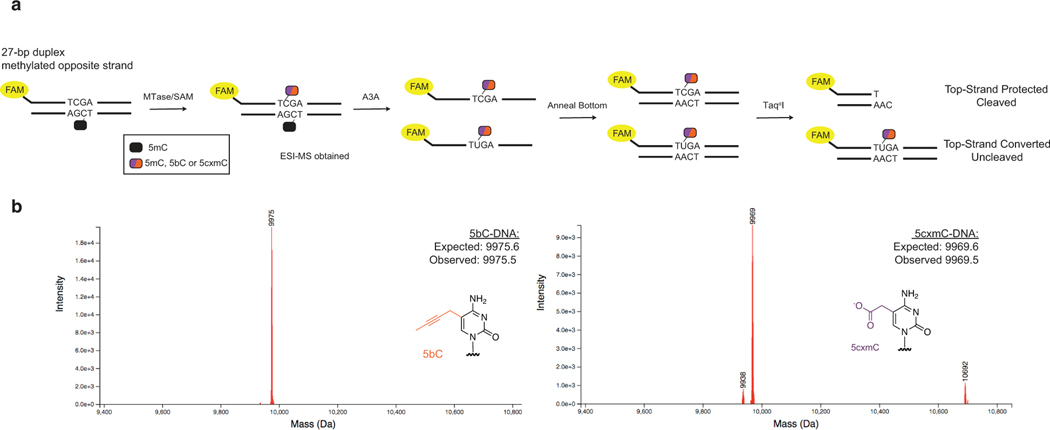
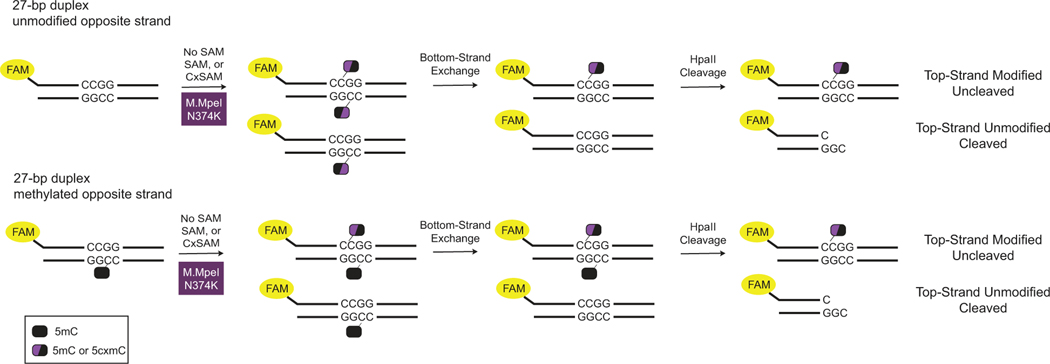
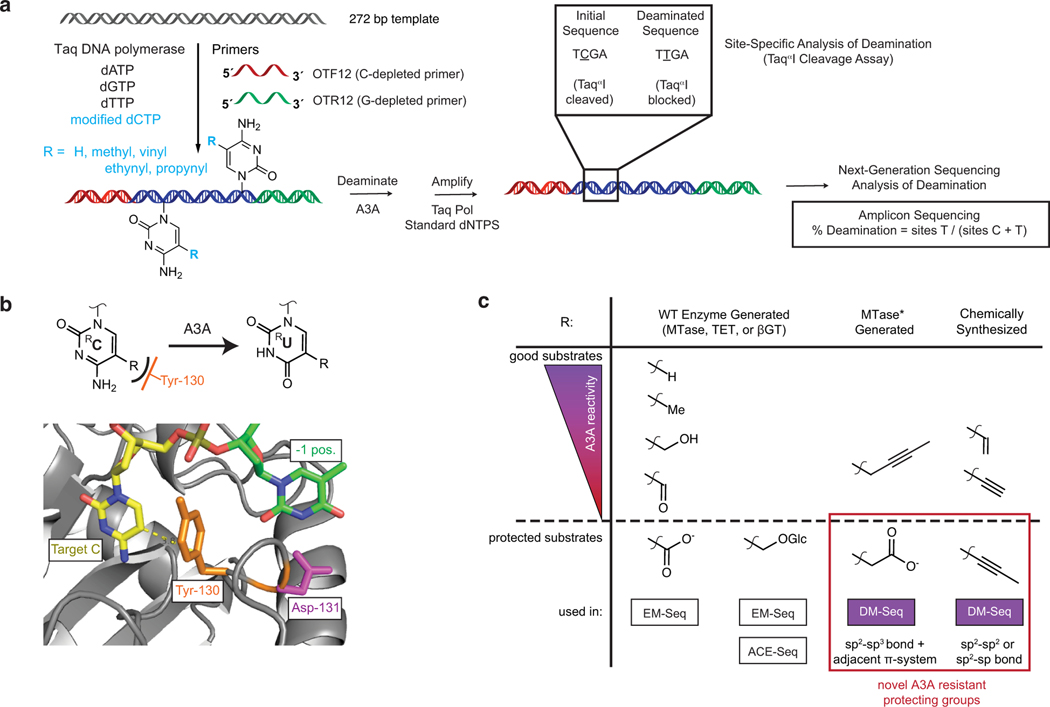
5-methylcytosine (5mC) is the most important DNA modification in mammalian genomes. The ideal method for 5mC localization would be both nondestructive of DNA and direct, without requiring inference based on detection of unmodified cytosines. Here we present direct methylation sequencing (DM-Seq), a bisulfite-free method for profiling 5mC at single-base resolution using nanogram quantities of DNA. DM-Seq employs two key DNA-modifying enzymes: a neomorphic DNA methyltransferase and a DNA deaminase capable of precise discrimination between cytosine modification states. Coupling these activities with deaminase-resistant adapters enables accurate detection of only 5mC via a C-to-T transition in sequencing. By comparison, we uncover a PCR-related underdetection bias with the hybrid enzymatic-chemical TET-assisted pyridine borane sequencing approach. Importantly, we show that DM-Seq, unlike bisulfite sequencing, unmasks prognostically important CpGs in a clinical tumor sample by not confounding 5mC with 5-hydroxymethylcytosine. DM-Seq thus offers an all-enzymatic, nondestructive, faithful and direct method for the reading of 5mC alone.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
COMPETING INTERESTS
The University of Pennsylvania has patents pending for CxMTase enzymes, DNA deaminase-resistant adapters and the DM-Seq pipeline. R.M.K. has served as a scientific advisory board member for Cambridge Epigenetix (CEGX). W.G. is an employee of CEGX and T.W. was supported by a fellowship from CEGX. A.D. and N.D. are employees of Integrated DNA Technologies, Inc. The remaining authors declare no competing interests.
Figures
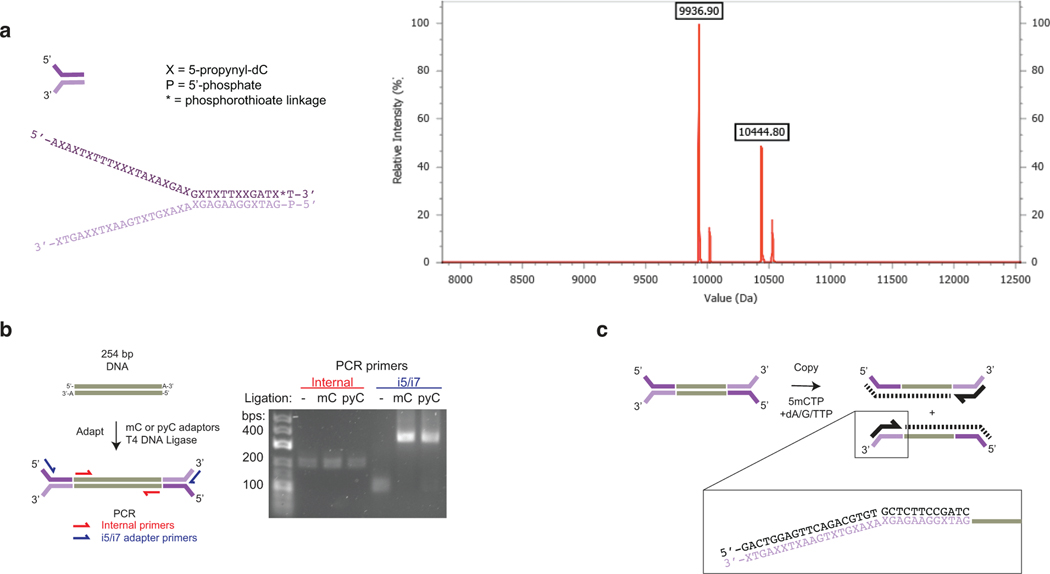
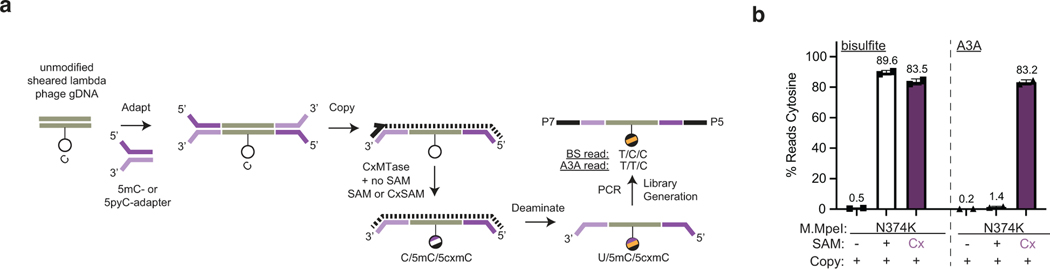
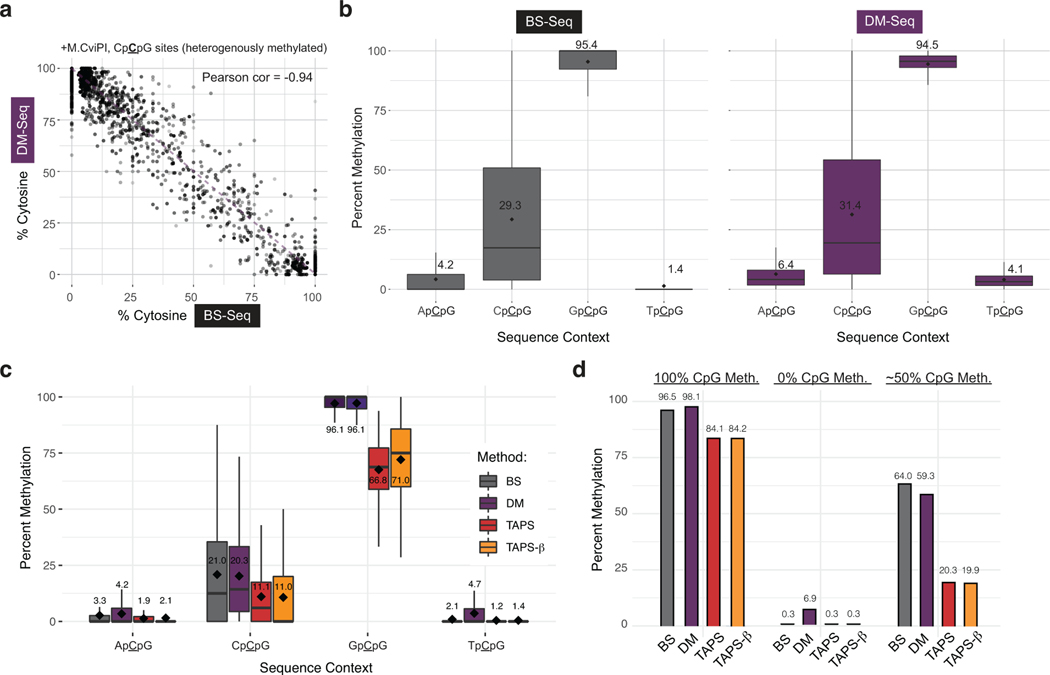
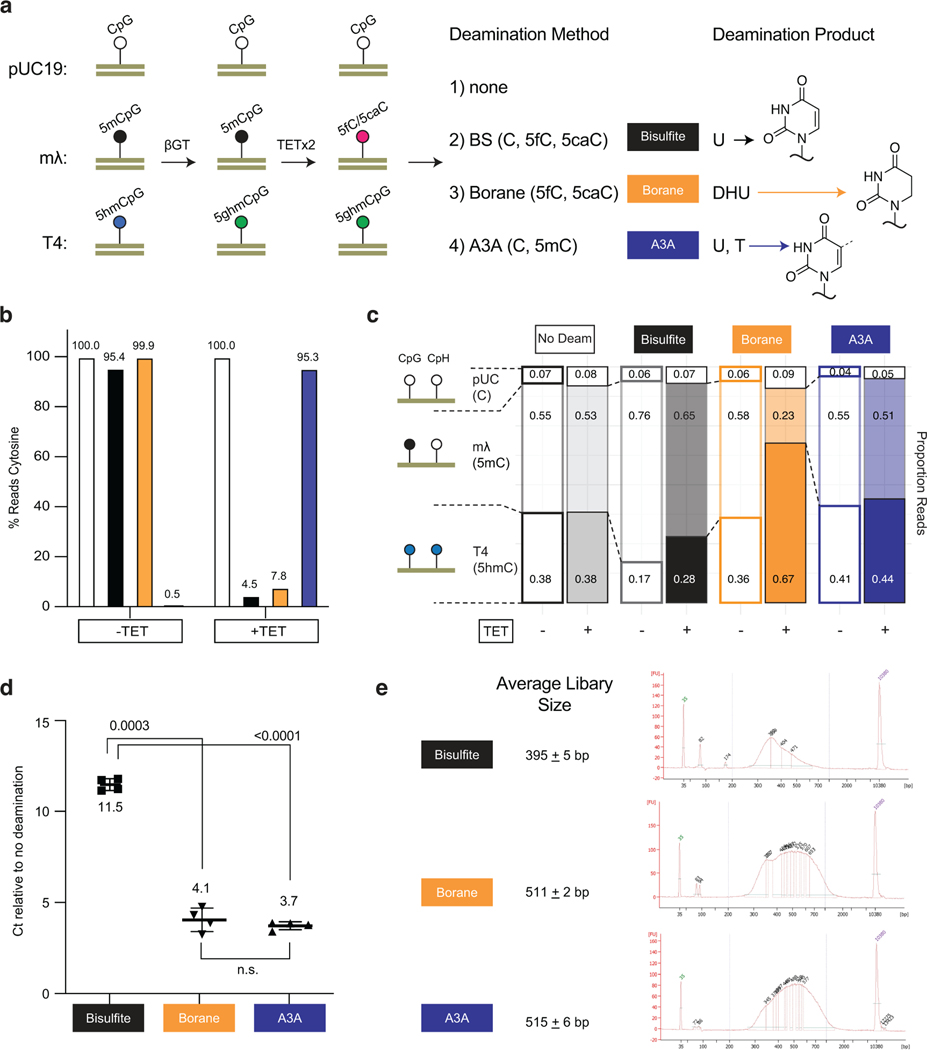
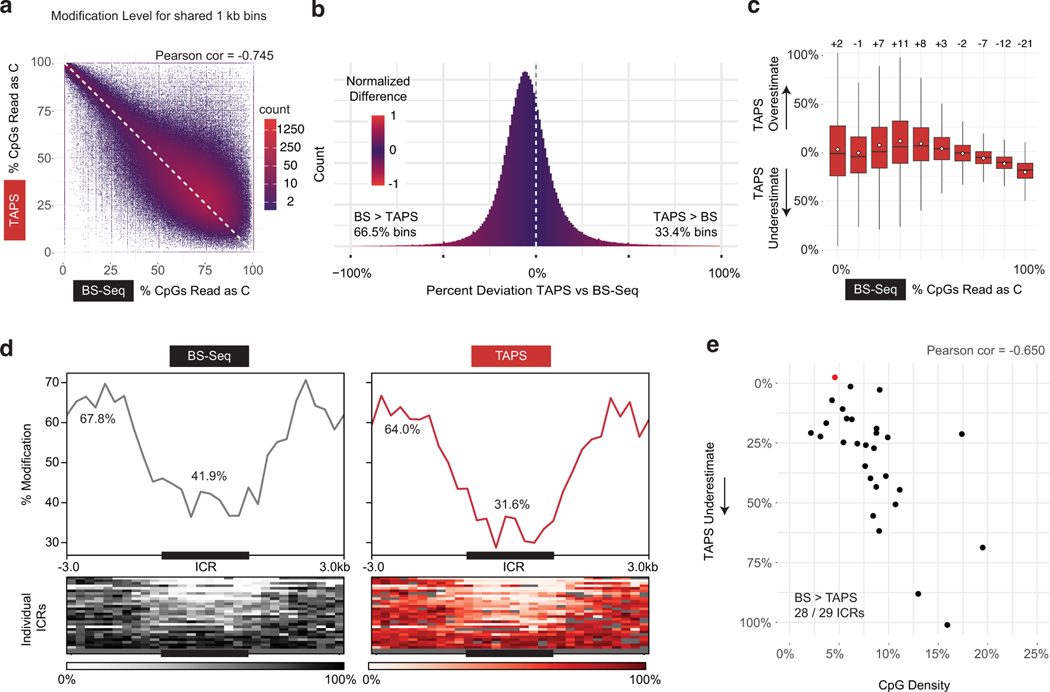
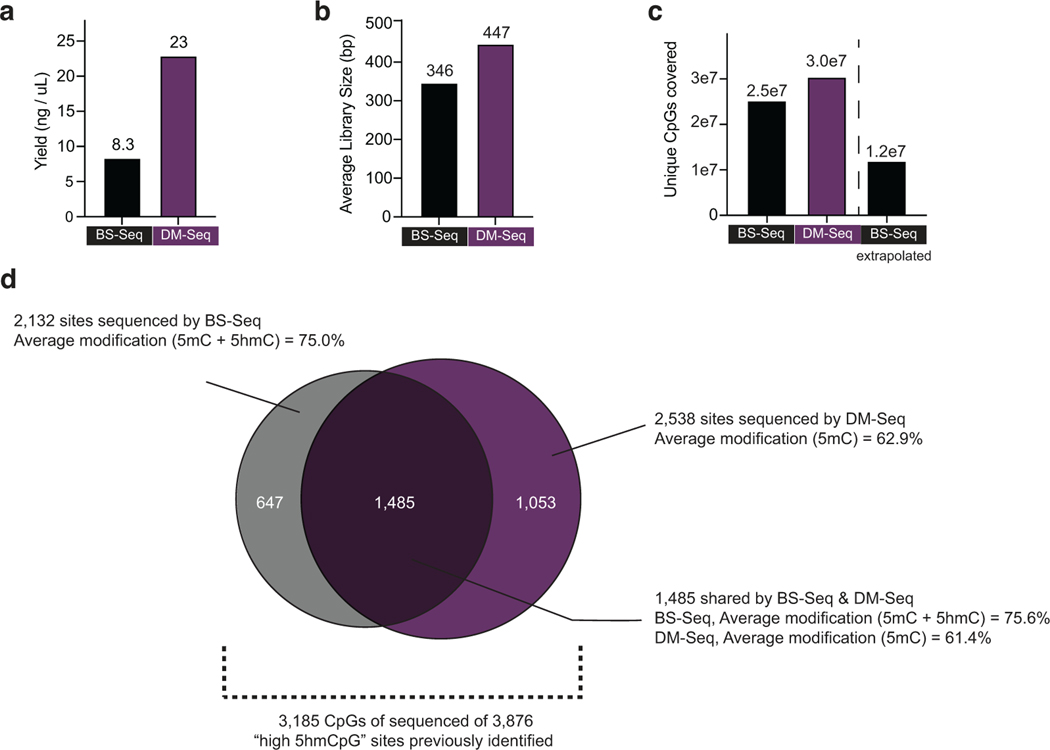
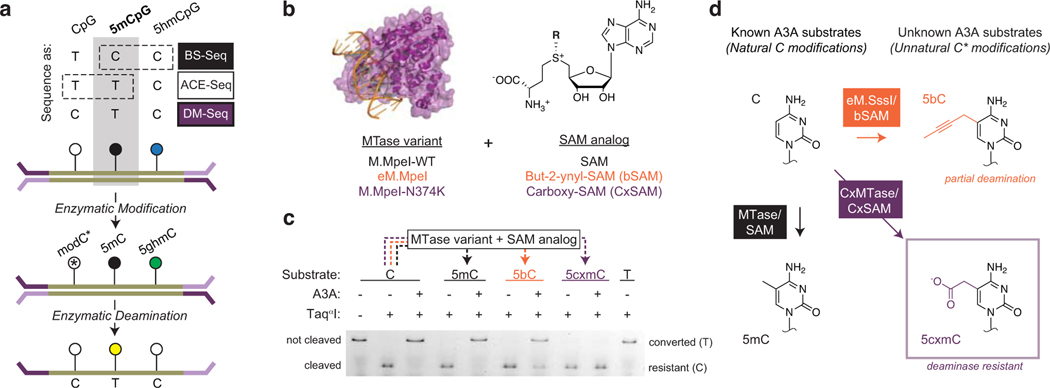
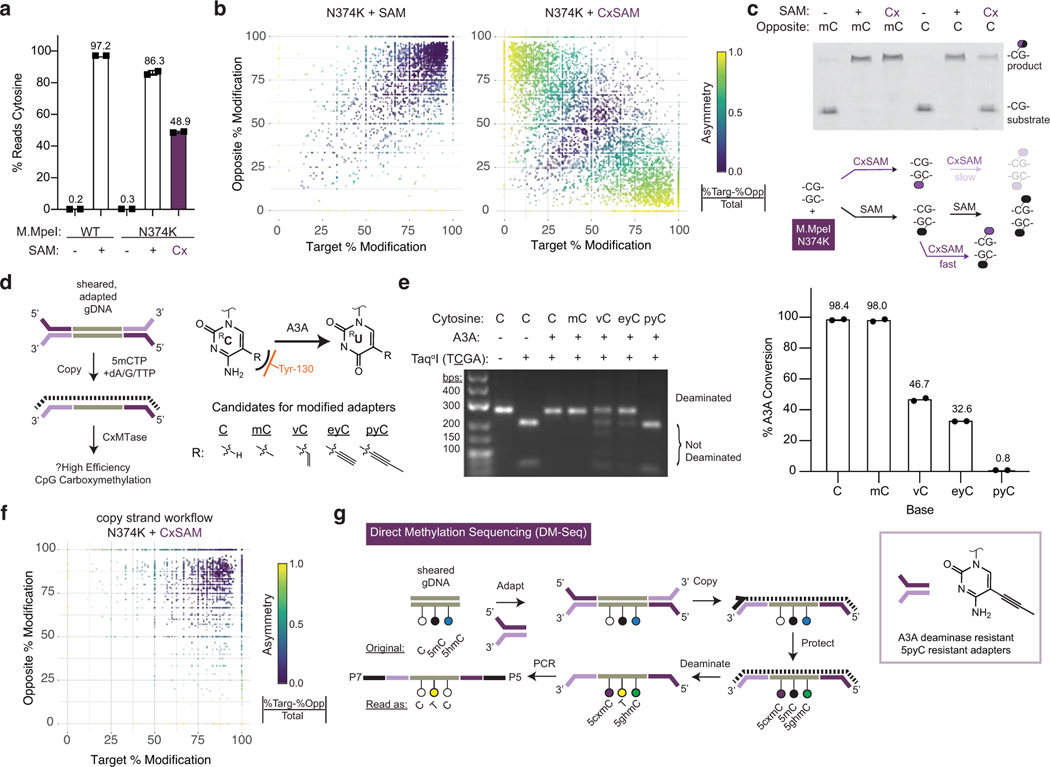
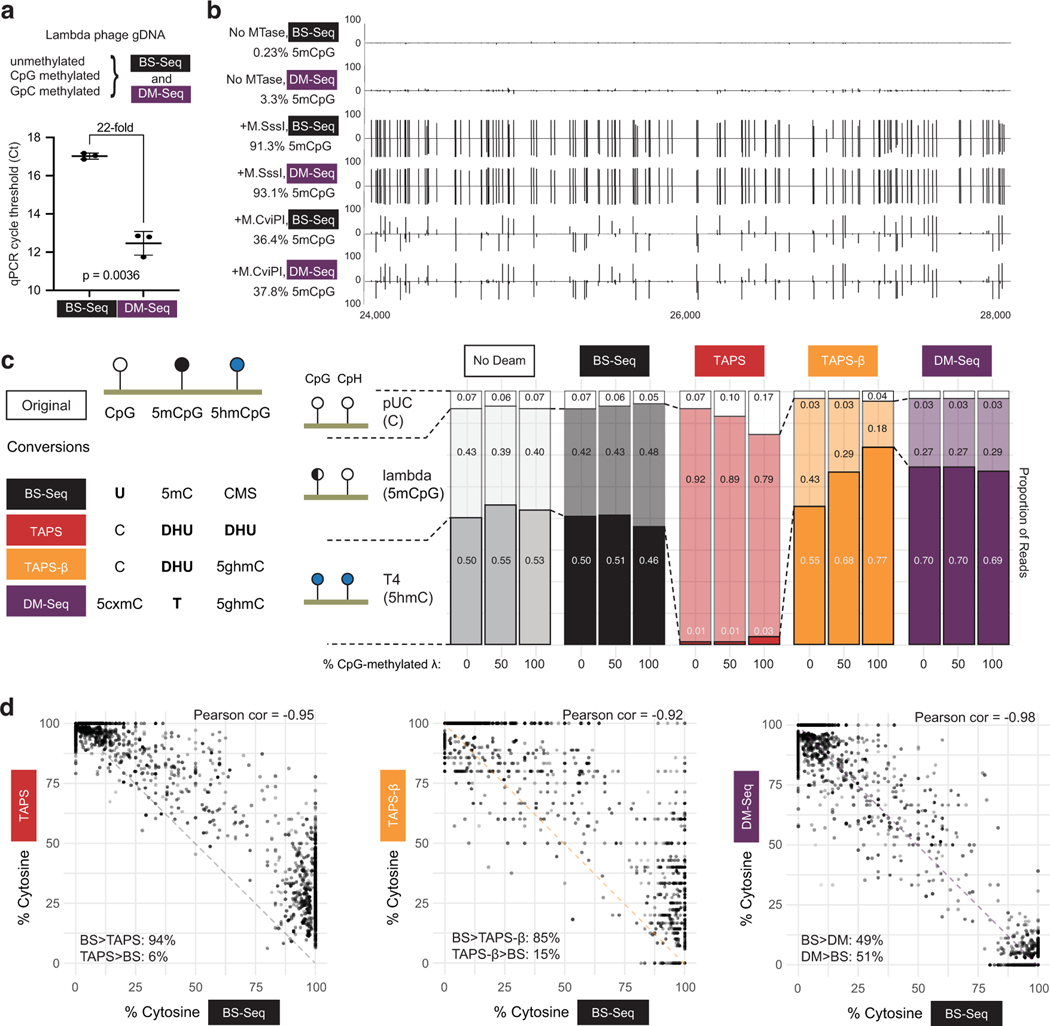
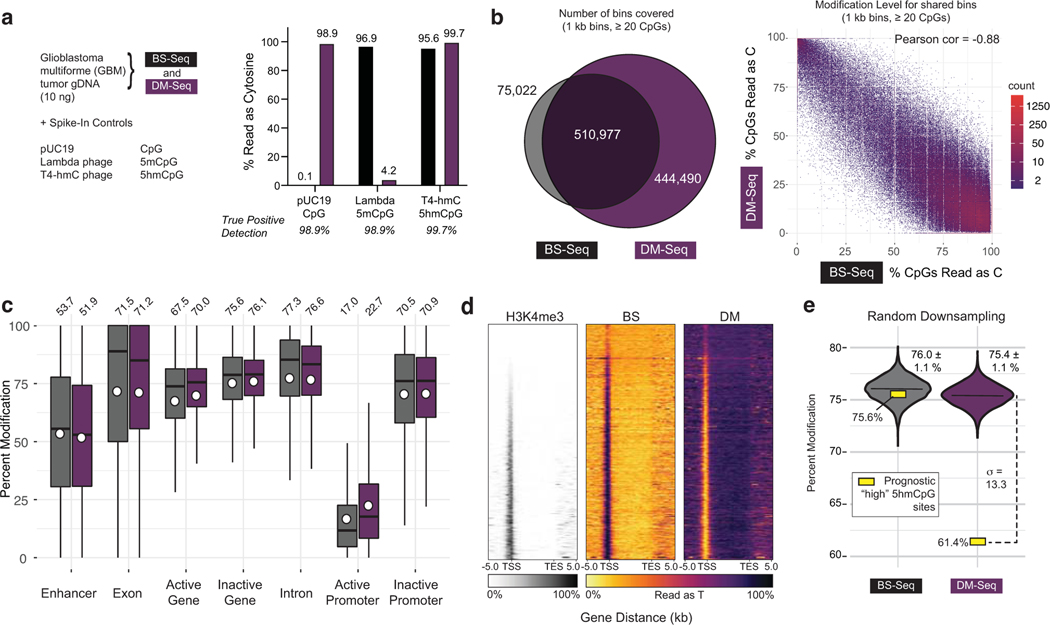
Comment in
-
Direct methylation sequencing.Nat Chem Biol. 2023 Aug;19(8):932-933. doi: 10.1038/s41589-023-01356-9. Nat Chem Biol. 2023. PMID: 37322154 No abstract available.
References
-
- Schubeler D. Function and information content of DNA methylation. Nature 517, 321–326 (2015). - PubMed
-
- Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T, Kis O, Zhao Z, Spreafico A, Medina TDS, Wang Y, Roulois D, Ettayebi I, Chen Z, Chow S, Murphy T, Arruda A, O’Kane GM, Liu J, Mansour M, McPherson JD, O’Brien C, Leighl N, Bedard PL, Fleshner N, Liu G, Minden MD, Gallinger S, Goldenberg A, Pugh TJ, Hoffman MM, Bratman SV, Hung RJ & De Carvalho DD Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018). - PubMed
-
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, Zhang J, Kandoth C, Akbani R, Shen H, Omberg L, Chu A, Margolin AA, Van’t Veer LJ, Lopez-Bigas N, Laird PW, Raphael BJ, Ding L, Robertson AG, Byers LA, Mills GB, Weinstein JN, Van Waes C, Chen Z, Collisson EA, Cancer Genome Atlas Research Network, Benz CC, Perou CM & Stuart JM Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014). - PMC - PubMed
METHODS ONLY REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
