

ReporTree: a surveillance-oriented tool to strengthen the linkage between pathogen genetic clusters and epidemiological data
- PMID: 37322495
- PMCID: PMC10273728
- DOI: 10.1186/s13073-023-01196-1
ReporTree: a surveillance-oriented tool to strengthen the linkage between pathogen genetic clusters and epidemiological data
Abstract
Background: Genomics-informed pathogen surveillance strengthens public health decision-making, playing an important role in infectious diseases' prevention and control. A pivotal outcome of genomics surveillance is the identification of pathogen genetic clusters and their characterization in terms of geotemporal spread or linkage to clinical and demographic data. This task often consists of the visual exploration of (large) phylogenetic trees and associated metadata, being time-consuming and difficult to reproduce.
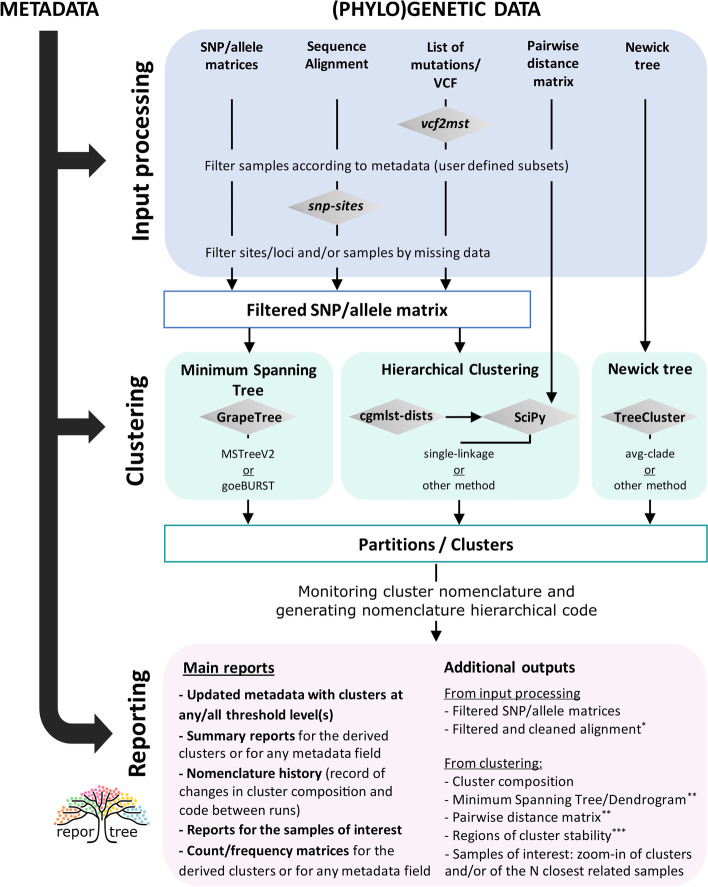
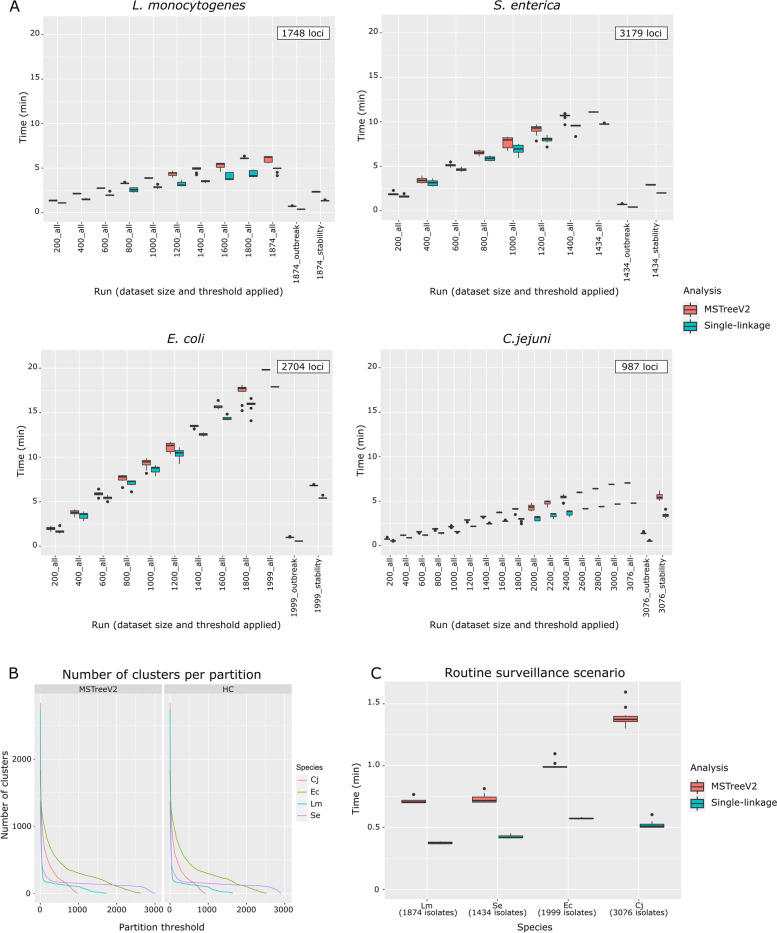
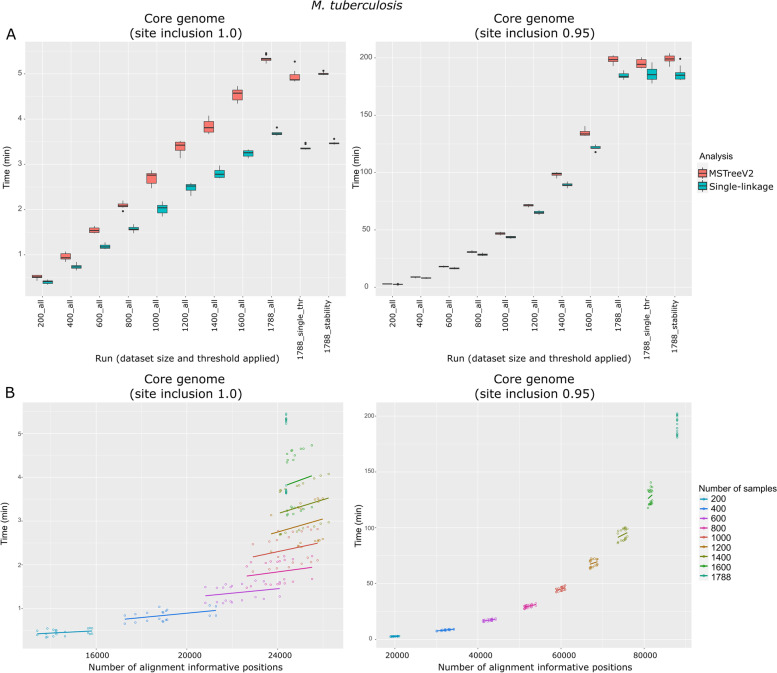
Results: We developed ReporTree, a flexible bioinformatics pipeline that allows diving into the complexity of pathogen diversity to rapidly identify genetic clusters at any (or all) distance threshold(s) or cluster stability regions and to generate surveillance-oriented reports based on the available metadata, such as timespan, geography, or vaccination/clinical status. ReporTree is able to maintain cluster nomenclature in subsequent analyses and to generate a nomenclature code combining cluster information at different hierarchical levels, thus facilitating the active surveillance of clusters of interest. By handling several input formats and clustering methods, ReporTree is applicable to multiple pathogens, constituting a flexible resource that can be smoothly deployed in routine surveillance bioinformatics workflows with negligible computational and time costs. This is demonstrated through a comprehensive benchmarking of (i) the cg/wgMLST workflow with large datasets of four foodborne bacterial pathogens and (ii) the alignment-based SNP workflow with a large dataset of Mycobacterium tuberculosis. To further validate this tool, we reproduced a previous large-scale study on Neisseria gonorrhoeae, demonstrating how ReporTree is able to rapidly identify the main species genogroups and characterize them with key surveillance metadata, such as antibiotic resistance data. By providing examples for SARS-CoV-2 and the foodborne bacterial pathogen Listeria monocytogenes, we show how this tool is currently a useful asset in genomics-informed routine surveillance and outbreak detection of a wide variety of species.
Conclusions: In summary, ReporTree is a pan-pathogen tool for automated and reproducible identification and characterization of genetic clusters that contributes to a sustainable and efficient public health genomics-informed pathogen surveillance. ReporTree is implemented in python 3.8 and is freely available at https://github.com/insapathogenomics/ReporTree .
Keywords: Automated pipeline; Genetic clustering; Genomic surveillance; Public health; ReporTree.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
