

Mutual information networks reveal evolutionary relationships within the influenza A virus polymerase
- PMID: 37325086
- PMCID: PMC10263469
- DOI: 10.1093/ve/vead037
Mutual information networks reveal evolutionary relationships within the influenza A virus polymerase
Abstract
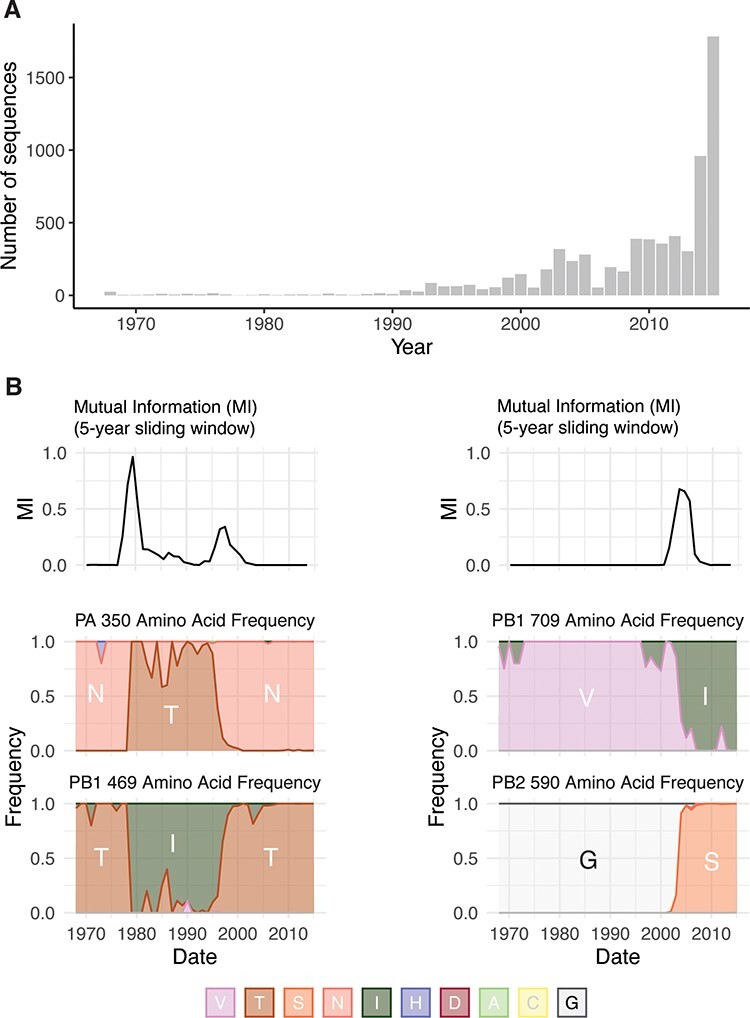
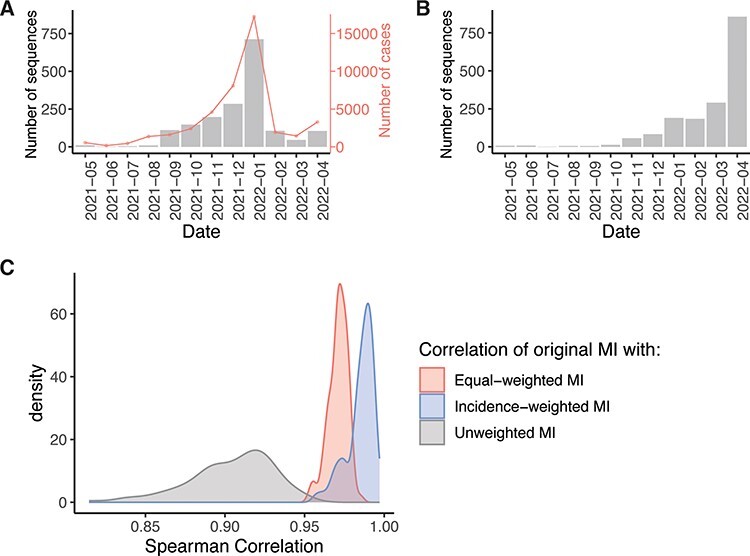
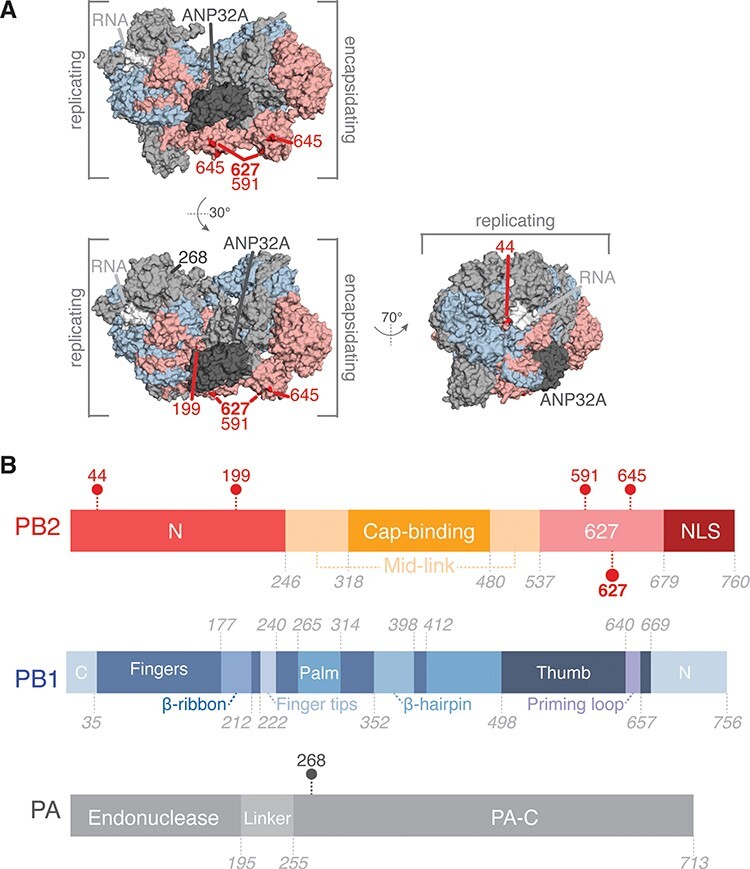
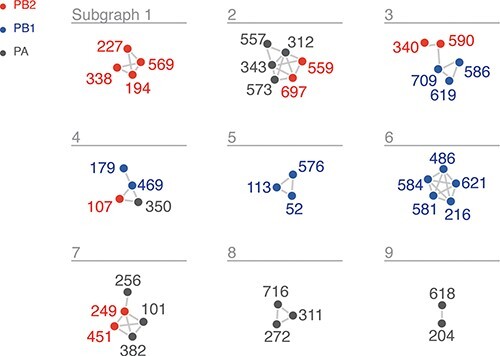
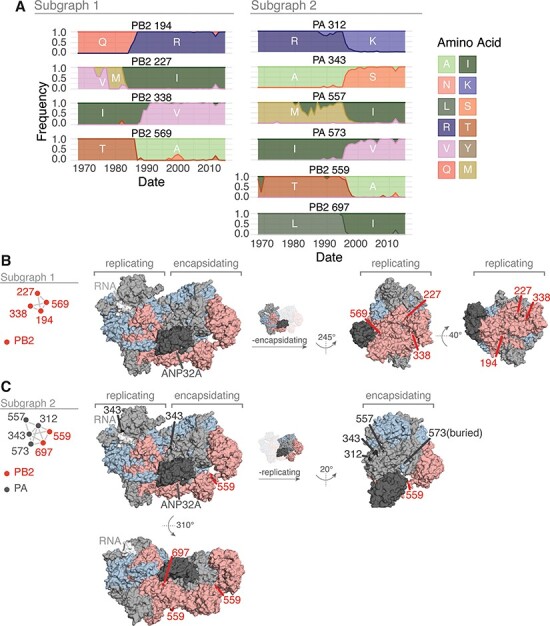
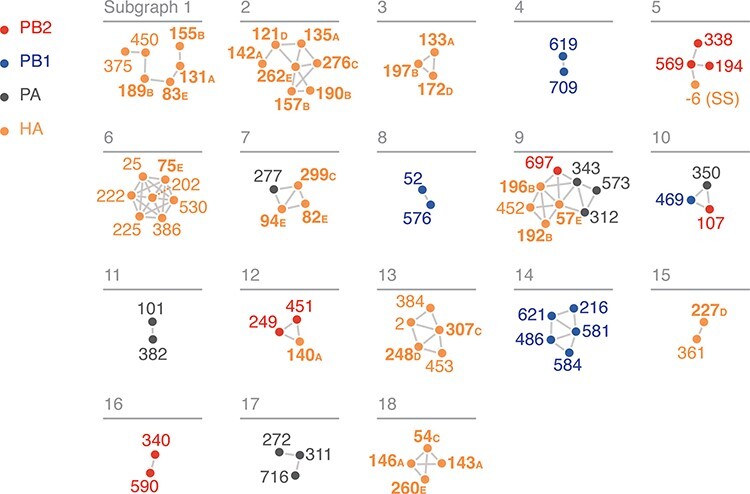
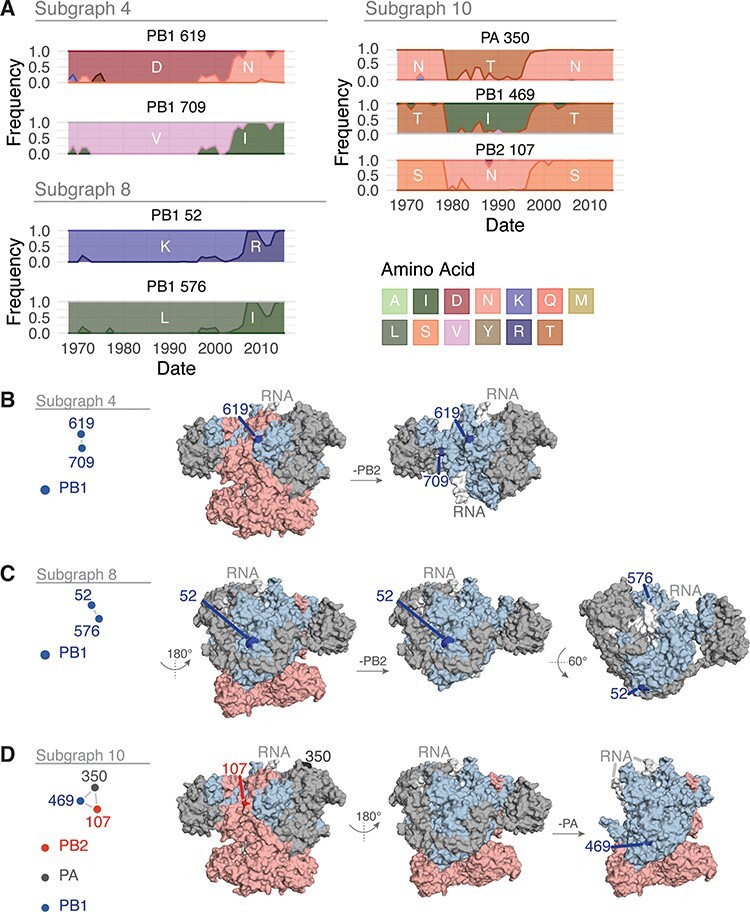
The influenza A virus (IAV) RNA polymerase is an essential driver of IAV evolution. Mutations that the polymerase introduces into viral genome segments during replication are the ultimate source of genetic variation, including within the three subunits of the IAV polymerase (polymerase basic protein 2, polymerase basic protein 1, and polymerase acidic protein). Evolutionary analysis of the IAV polymerase is complicated, because changes in mutation rate, replication speed, and drug resistance involve epistatic interactions among its subunits. In order to study the evolution of the human seasonal H3N2 polymerase since the 1968 pandemic, we identified pairwise evolutionary relationships among ∼7000 H3N2 polymerase sequences using mutual information (MI), which measures the information gained about the identity of one residue when a second residue is known. To account for uneven sampling of viral sequences over time, we developed a weighted MI (wMI) metric and demonstrate that wMI outperforms raw MI through simulations using a well-sampled severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) dataset. We then constructed wMI networks of the H3N2 polymerase to extend the inherently pairwise wMI statistic to encompass relationships among larger groups of residues. We included hemagglutinin (HA) in the wMI network to distinguish between functional wMI relationships within the polymerase and those potentially due to hitch-hiking on antigenic changes in HA. The wMI networks reveal coevolutionary relationships among residues with roles in replication and encapsidation. Inclusion of HA highlighted polymerase-only subgraphs containing residues with roles in the enzymatic functions of the polymerase and host adaptability. This work provides insight into the factors that drive and constrain the rapid evolution of influenza viruses.
Keywords: Influenza Avirus; RNA-dependent RNA polymerase; information theory; virus evolution.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
Update of
-
Mutual information networks reveal evolutionary relationships within the influenza A virus polymerase.bioRxiv [Preprint]. 2023 Feb 17:2023.02.16.528850. doi: 10.1101/2023.02.16.528850. bioRxiv. 2023. Update in: Virus Evol. 2023 May 27;9(1):vead037. doi: 10.1093/ve/vead037. PMID: 36824962 Free PMC article. Updated. Preprint.
References
-
- Chang W. et al. (2022), Shiny: Web Applicatoin Framework for R. <https://shiny.rstudio.com/> accessed 1 Jan 2021.
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
