

Efficiently quantifying DNA methylation for bulk- and single-cell bisulfite data
- PMID: 37326968
- PMCID: PMC10310462
- DOI: 10.1093/bioinformatics/btad386
Efficiently quantifying DNA methylation for bulk- and single-cell bisulfite data
Abstract
Motivation: DNA CpG methylation (CpGm) has proven to be a crucial epigenetic factor in the mammalian gene regulatory system. Assessment of DNA CpG methylation values via whole-genome bisulfite sequencing (WGBS) is, however, computationally extremely demanding.
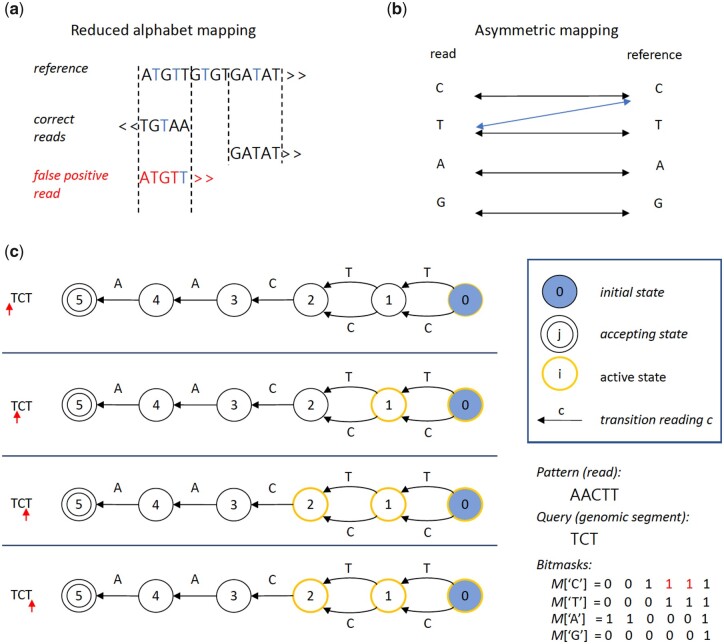
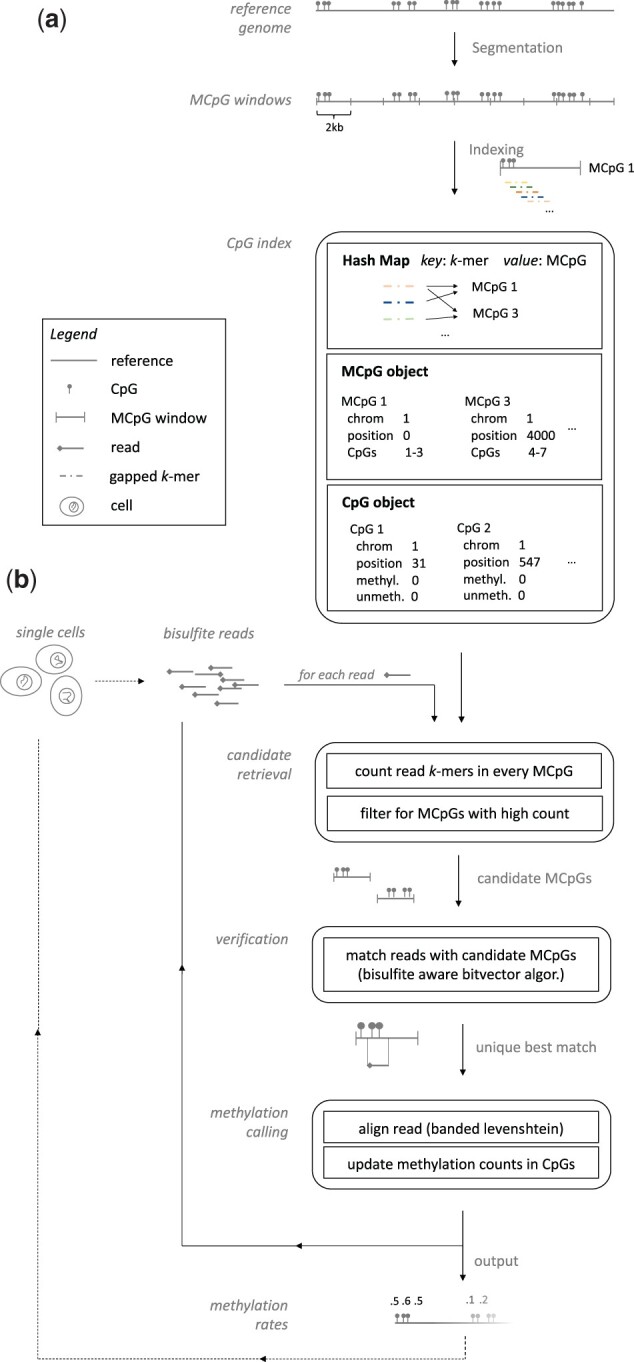
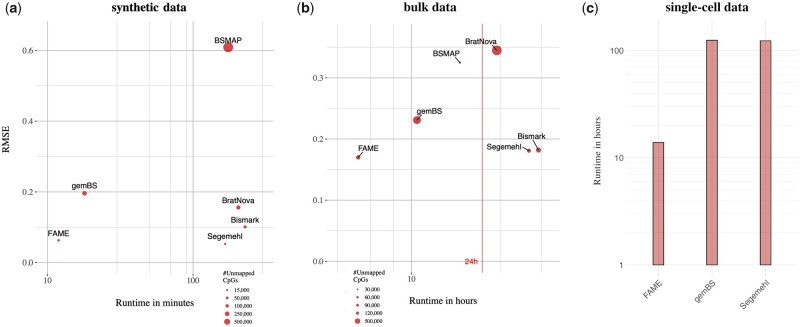
Results: We present FAst MEthylation calling (FAME), the first approach to quantify CpGm values directly from bulk or single-cell WGBS reads without intermediate output files. FAME is very fast but as accurate as standard methods, which first produce BS alignment files before computing CpGm values. We present experiments on bulk and single-cell bisulfite datasets in which we show that data analysis can be significantly sped-up and help addressing the current WGBS analysis bottleneck for large-scale datasets without compromising accuracy.
Availability and implementation: An implementation of FAME is open source and licensed under GPL-3.0 at https://github.com/FischerJo/FAME.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures

Similar articles
-
MethylStar: A fast and robust pre-processing pipeline for bulk or single-cell whole-genome bisulfite sequencing data.BMC Genomics. 2020 Jul 13;21(1):479. doi: 10.1186/s12864-020-06886-3. BMC Genomics. 2020. PMID: 32660416 Free PMC article.
-
BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data.BMC Genomics. 2013 Nov 10;14:774. doi: 10.1186/1471-2164-14-774. BMC Genomics. 2013. PMID: 24206606 Free PMC article.
-
A comprehensive evaluation of alignment software for reduced representation bisulfite sequencing data.Bioinformatics. 2018 Aug 15;34(16):2715-2723. doi: 10.1093/bioinformatics/bty174. Bioinformatics. 2018. PMID: 29579198
-
Methodological aspects of whole-genome bisulfite sequencing analysis.Brief Bioinform. 2015 May;16(3):369-79. doi: 10.1093/bib/bbu016. Epub 2014 May 27. Brief Bioinform. 2015. PMID: 24867940 Review.
-
Strategies for analyzing bisulfite sequencing data.J Biotechnol. 2017 Nov 10;261:105-115. doi: 10.1016/j.jbiotec.2017.08.007. Epub 2017 Aug 16. J Biotechnol. 2017. PMID: 28822795 Review.
References
-
- Baeza-Yates R, Gonnet GH.. A new approach to text searching. Commun ACM 1992;35:74–82.
-
- Bray NL, Pimentel H, Melsted P. et al. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34:525–7. - PubMed
