

The CD58-CD2 axis is co-regulated with PD-L1 via CMTM6 and shapes anti-tumor immunity
- PMID: 37327789
- PMCID: PMC10524902
- DOI: 10.1016/j.ccell.2023.05.014
The CD58-CD2 axis is co-regulated with PD-L1 via CMTM6 and shapes anti-tumor immunity
Abstract
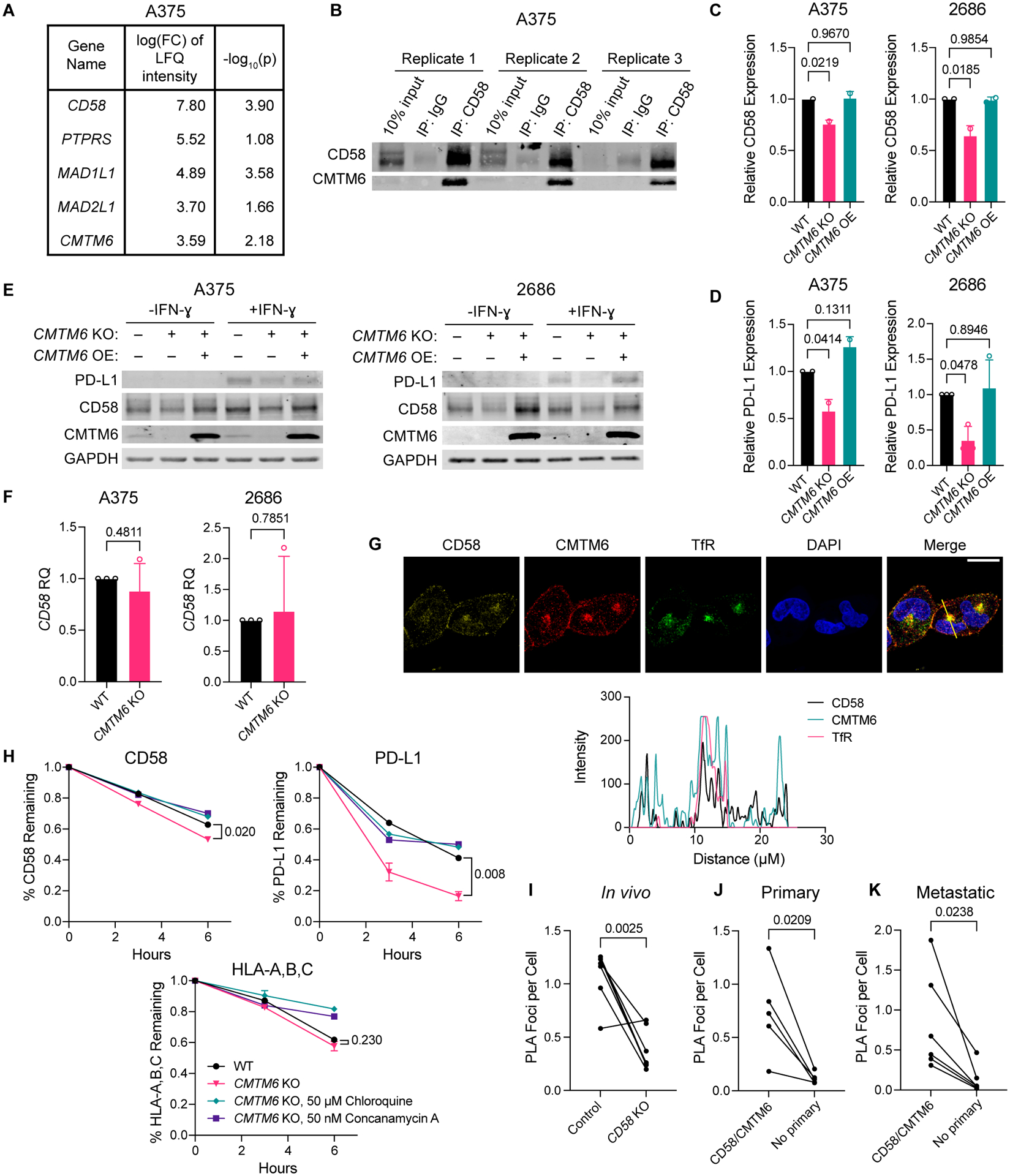
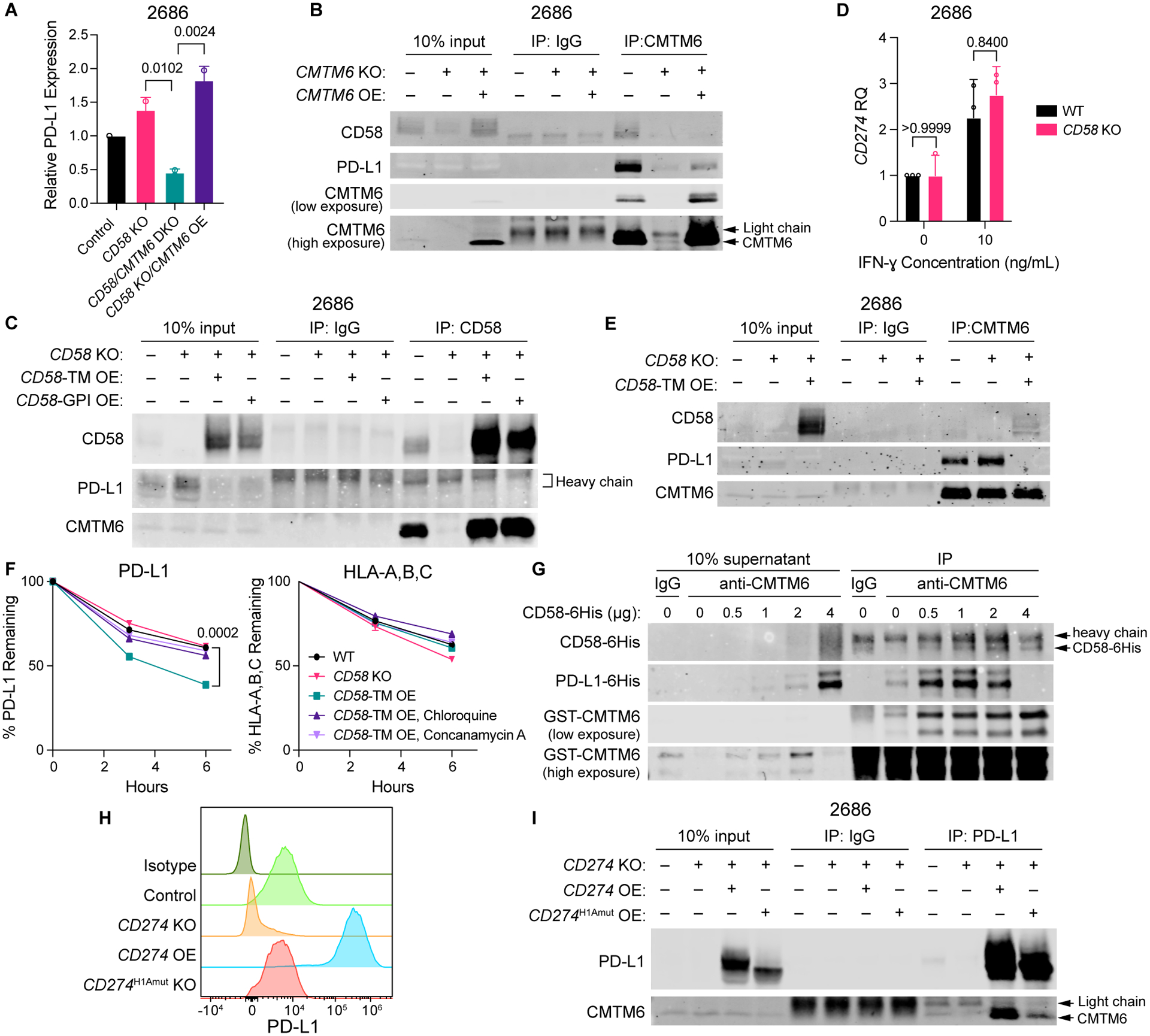
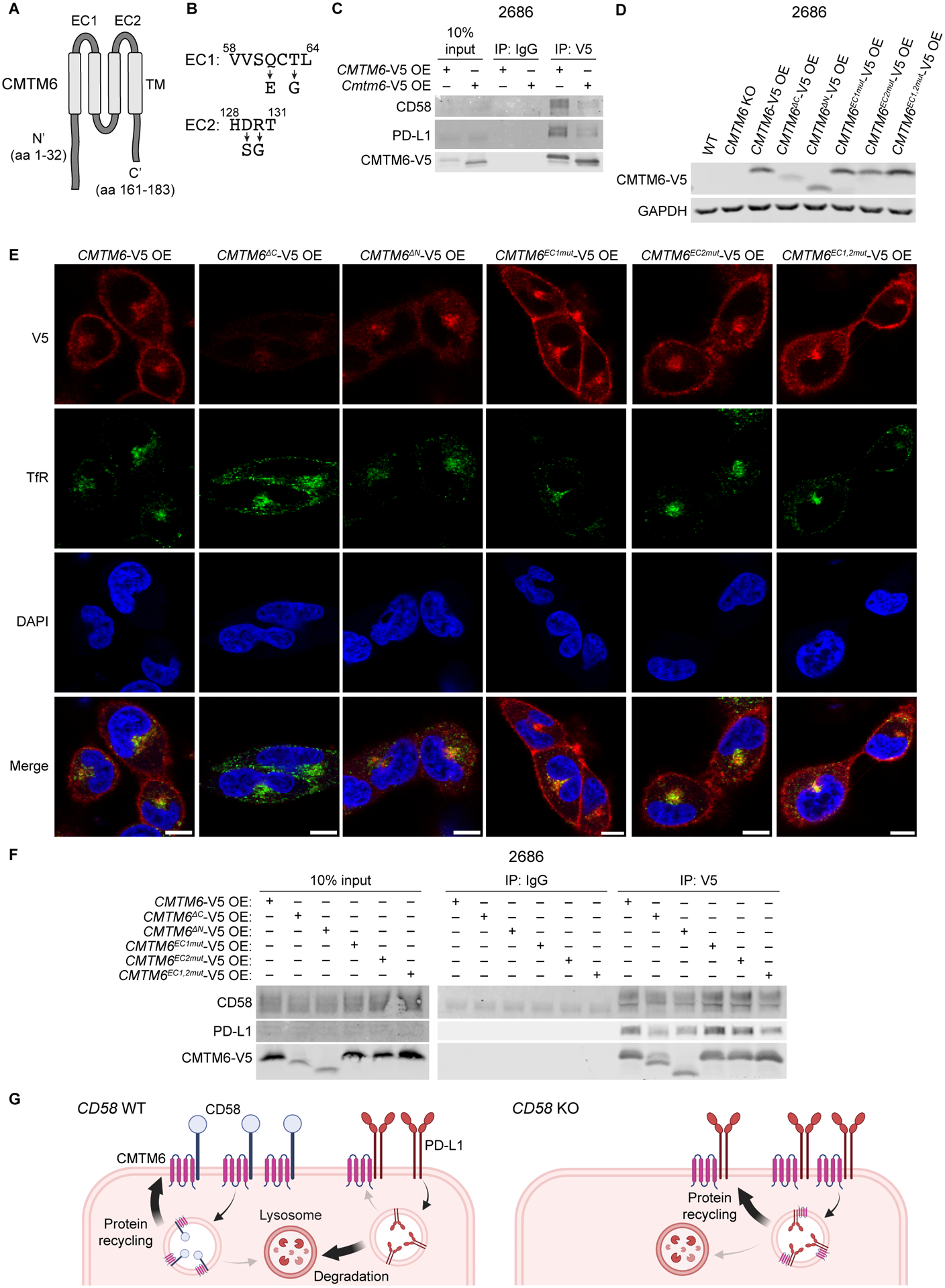
The cell-autonomous balance of immune-inhibitory and -stimulatory signals is a critical process in cancer immune evasion. Using patient-derived co-cultures, humanized mouse models, and single-cell RNA-sequencing of patient melanomas biopsied before and on immune checkpoint blockade, we find that intact cancer cell-intrinsic expression of CD58 and ligation to CD2 is required for anti-tumor immunity and is predictive of treatment response. Defects in this axis promote immune evasion through diminished T cell activation, impaired intratumoral T cell infiltration and proliferation, and concurrently increased PD-L1 protein stabilization. Through CRISPR-Cas9 and proteomics screens, we identify and validate CMTM6 as critical for CD58 stability and upregulation of PD-L1 upon CD58 loss. Competition between CD58 and PD-L1 for CMTM6 binding determines their rate of endosomal recycling over lysosomal degradation. Overall, we describe an underappreciated yet critical axis of cancer immunity and provide a molecular basis for how cancer cells balance immune inhibitory and stimulatory cues.
Keywords: CD2; CD58; CRISPR-Cas9 screen; PDL1; balance of co-inhibitory/co-stimulator; cancer immune evasion; cancer immunology; cancer immunotherapy; immune checkpoint blockade; mass spec screen; resistance to immune checkpoint blockade; single-cell RNA-sequencing.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.I. is a consultant for or received honoraria from Volastra Therapeutics, Johnson & Johnson/Janssen, Novartis, Eisai, AstraZeneca and Merck, and has received research funding to Columbia University from Agenus, Alkermes, Arcus Biosciences, Checkmate Pharmaceuticals, Compugen, Immunocore, and Synthekine. K.W.W. serves on the scientific advisory board of T-Scan Therapeutics, SQZ Biotech, Nextechinvest and receives sponsored research funding from Novartis. He is a co-founder of Immunitas, a biotech company. None of these represent a conflict of interest pertaining to the presented work. P.H., J.C.M., and B.I. filed a patent pertaining to the presented work.
Figures
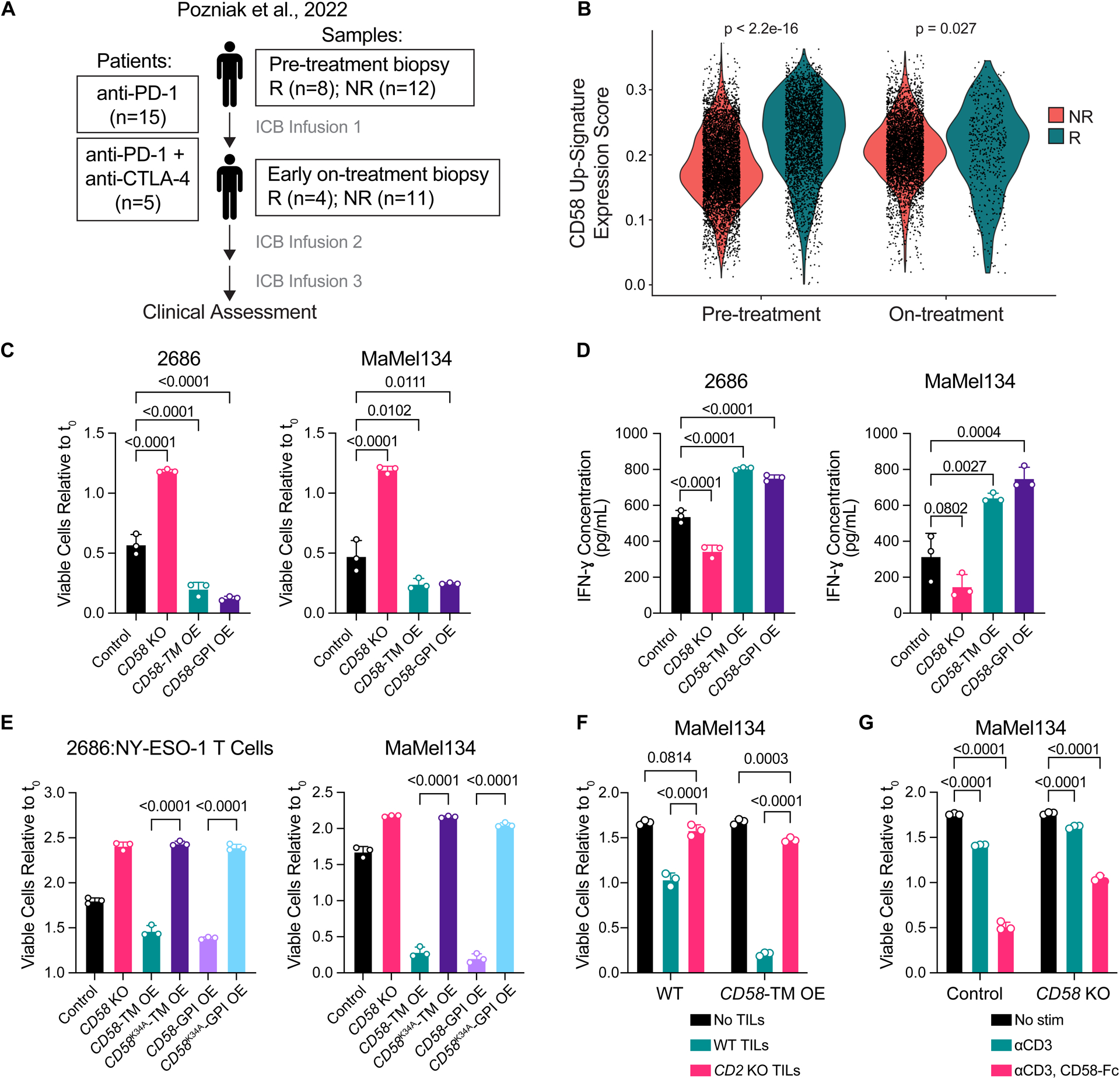
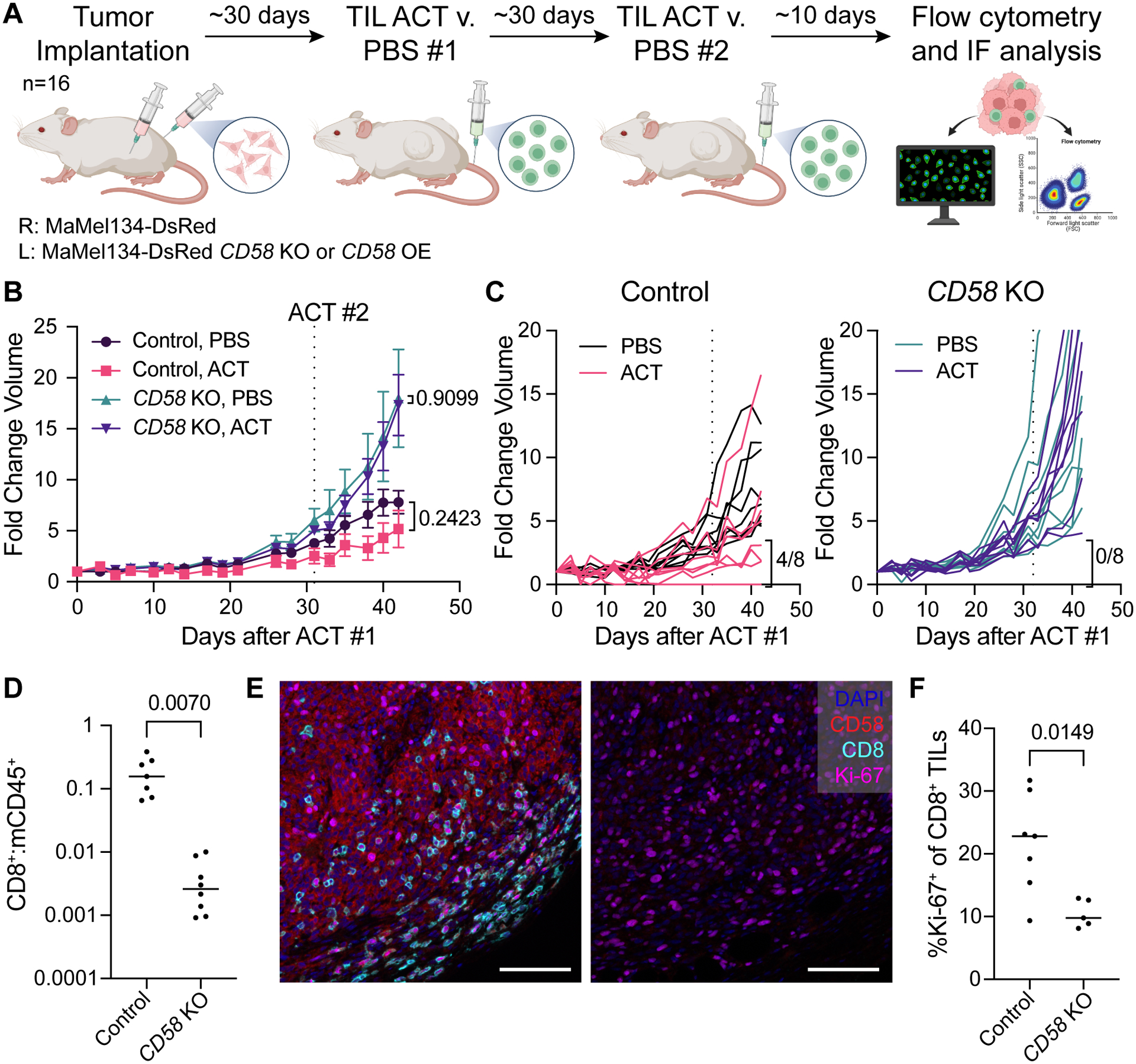
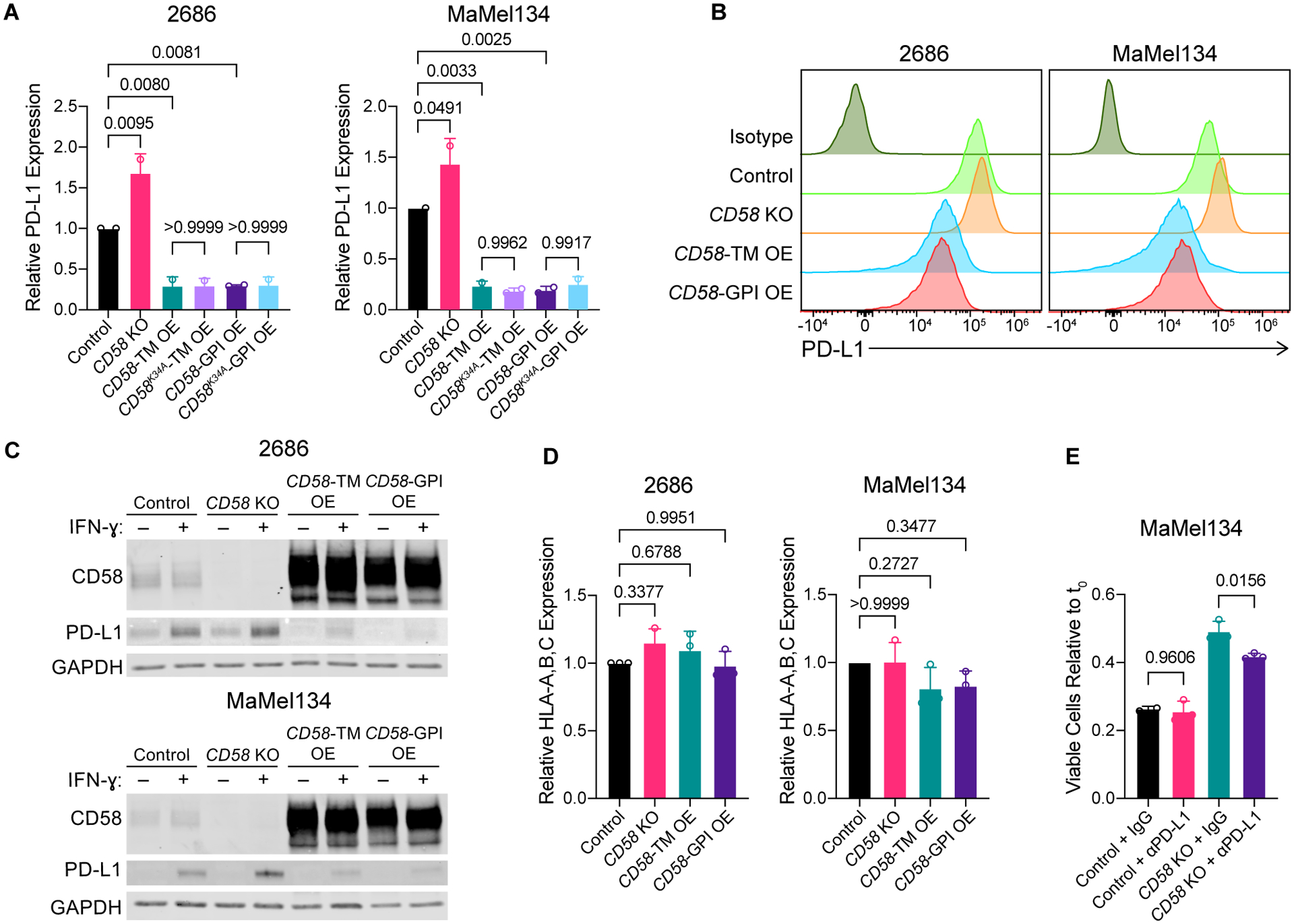
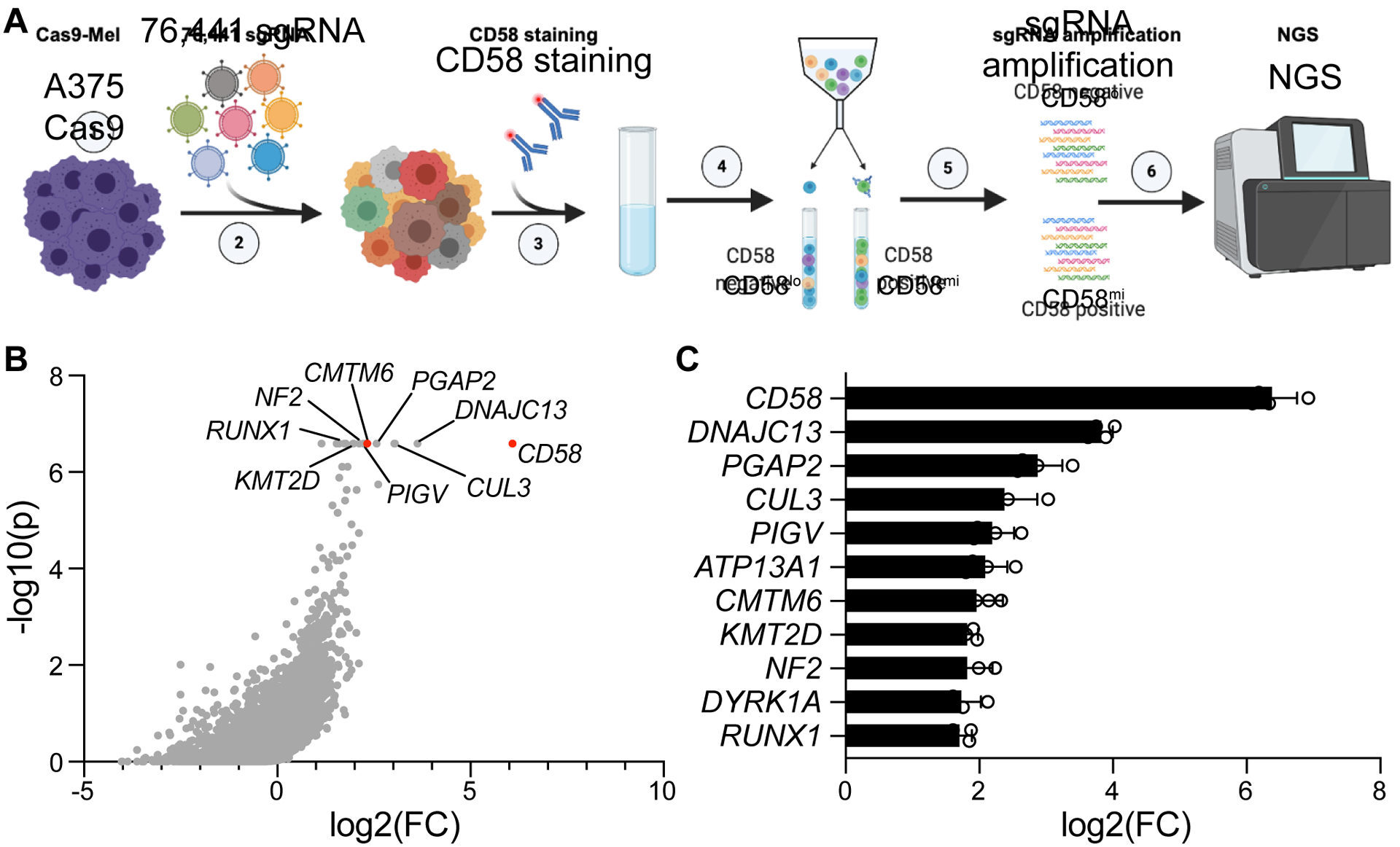
References
-
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph R, Weber JS, et al. (2019). Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Annals of Oncology: Official Journal of the European Society for Medical Oncology 30, 582–588. 10.1093/annonc/mdz011. - DOI - PMC - PubMed
-
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England Journal of Medicine 381, 1535–1546. 10.1056/NEJMoa1910836. - DOI - PubMed
-
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nature Communications 8, 1136. 10.1038/s41467017-01062-w. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
