

Evaluation of a genetic risk score computed using human chromosomal-scale length variation to predict breast cancer
- PMID: 37328908
- PMCID: PMC10273758
- DOI: 10.1186/s40246-023-00482-8
Evaluation of a genetic risk score computed using human chromosomal-scale length variation to predict breast cancer
Abstract
Introduction: The ability to accurately predict whether a woman will develop breast cancer later in her life, should reduce the number of breast cancer deaths. Different predictive models exist for breast cancer based on family history, BRCA status, and SNP analysis. The best of these models has an accuracy (area under the receiver operating characteristic curve, AUC) of about 0.65. We have developed computational methods to characterize a genome by a small set of numbers that represent the length of segments of the chromosomes, called chromosomal-scale length variation (CSLV).
Methods: We built machine learning models to differentiate between women who had breast cancer and women who did not based on their CSLV characterization. We applied this procedure to two different datasets: the UK Biobank (1534 women with breast cancer and 4391 women who did not) and the Cancer Genome Atlas (TCGA) 874 with breast cancer and 3381 without.
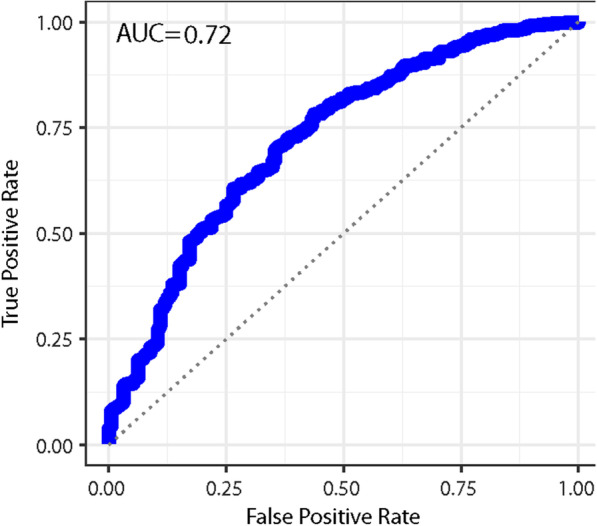
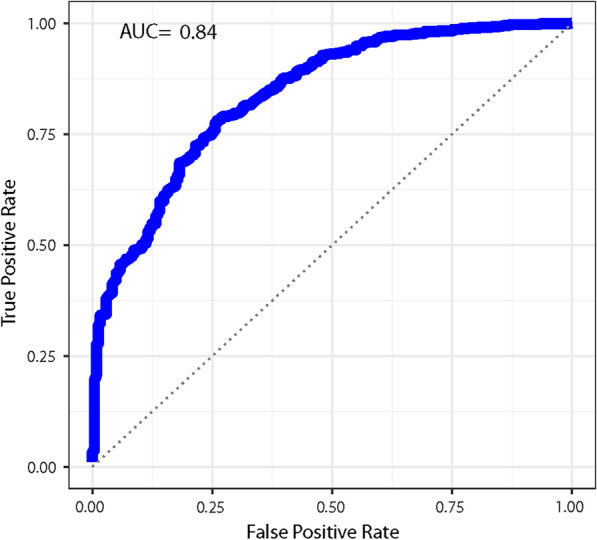
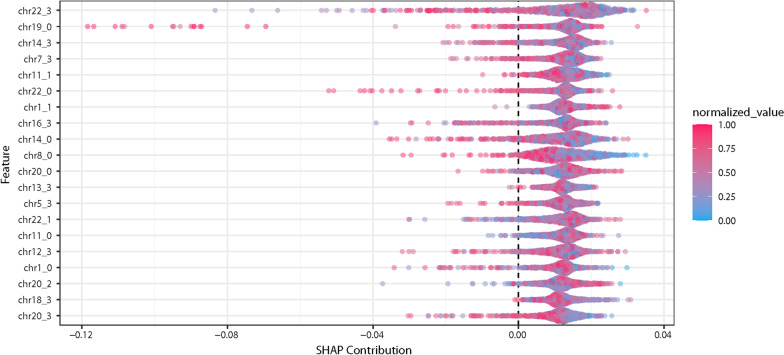
Results: We found a machine learning model that could predict breast cancer with an AUC of 0.836 95% CI (0.830.0.843) in the UK Biobank data. Using a similar approach with the TCGA data, we obtained a model with an AUC of 0.704 95% CI (0.702, 0.706). Variable importance analysis indicated that no single chromosomal region was responsible for significant fraction of the model results.
Conclusion: In this retrospective study, chromosomal-scale length variation could effectively predict whether or not a woman enrolled in the UK Biobank study developed breast cancer.
Keywords: Breast cancer; Copy number variation; Germline; Machine learning; TCGA; UK biobank; h2o.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Improved breast cancer risk prediction using chromosomal-scale length variation.Hum Genomics. 2025 Jun 11;19(1):65. doi: 10.1186/s40246-025-00776-z. Hum Genomics. 2025. PMID: 40500782 Free PMC article.
-
Genetic risk score for ovarian cancer based on chromosomal-scale length variation.BioData Min. 2021 Mar 9;14(1):18. doi: 10.1186/s13040-021-00253-y. BioData Min. 2021. PMID: 33750420 Free PMC article.
-
A genetic risk score using human chromosomal-scale length variation can predict schizophrenia.Sci Rep. 2021 Sep 22;11(1):18866. doi: 10.1038/s41598-021-97983-0. Sci Rep. 2021. PMID: 34552103 Free PMC article.
-
A genetic risk score for glioblastoma multiforme based on copy number variations.Cancer Treat Res Commun. 2021;27:100352. doi: 10.1016/j.ctarc.2021.100352. Epub 2021 Mar 16. Cancer Treat Res Commun. 2021. PMID: 33756171
-
Machine learning with magnetic resonance imaging for prediction of response to neoadjuvant chemotherapy in breast cancer: A systematic review and meta-analysis.Eur J Radiol. 2022 May;150:110247. doi: 10.1016/j.ejrad.2022.110247. Epub 2022 Mar 10. Eur J Radiol. 2022. PMID: 35290910
Cited by
-
A compact encoding of the genome suitable for machine learning prediction of traits and genetic risk scores.BioData Min. 2025 Jun 19;18(1):44. doi: 10.1186/s13040-025-00459-4. BioData Min. 2025. PMID: 40537821 Free PMC article.
-
Improved breast cancer risk prediction using chromosomal-scale length variation.Hum Genomics. 2025 Jun 11;19(1):65. doi: 10.1186/s40246-025-00776-z. Hum Genomics. 2025. PMID: 40500782 Free PMC article.
-
A contemporary review of breast cancer risk factors and the role of artificial intelligence.Front Oncol. 2024 Apr 18;14:1356014. doi: 10.3389/fonc.2024.1356014. eCollection 2024. Front Oncol. 2024. PMID: 38699635 Free PMC article. Review.
References
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71. - PubMed
-
- Krontiras H, Farmer M, Whatley J. Breast cancer genetics and indications for prophylactic mastectomy. Surgical Clinics of North America. 2018. - PubMed
-
- Chlebowski RT, Anderson GL, Lane DS, Aragaki AK, Rohan T, Yasmeen S, et al. Predicting risk of breast cancer in postmenopausal women by hormone receptor status. J Natl Cancer Inst. 2007;99. - PubMed
