

Fragmentomic analysis of circulating tumor DNA-targeted cancer panels
- PMID: 37330052
- PMCID: PMC10527168
- DOI: 10.1016/j.annonc.2023.06.001
Fragmentomic analysis of circulating tumor DNA-targeted cancer panels
Abstract
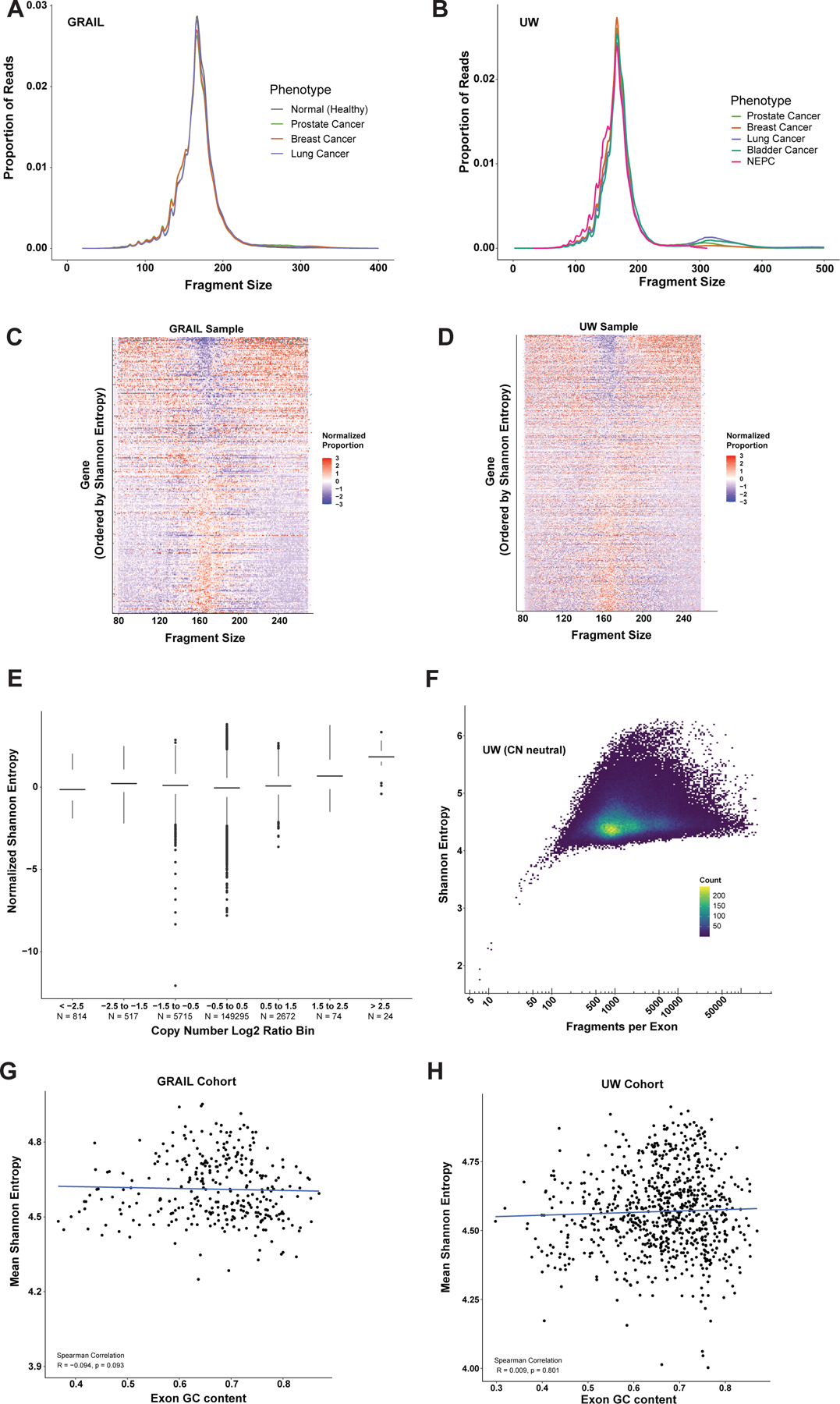
Background: The isolation of cell-free DNA (cfDNA) from the bloodstream can be used to detect and analyze somatic alterations in circulating tumor DNA (ctDNA), and multiple cfDNA-targeted sequencing panels are now commercially available for Food and Drug Administration (FDA)-approved biomarker indications to guide treatment. More recently, cfDNA fragmentation patterns have emerged as a tool to infer epigenomic and transcriptomic information. However, most of these analyses used whole-genome sequencing, which is insufficient to identify FDA-approved biomarker indications in a cost-effective manner.
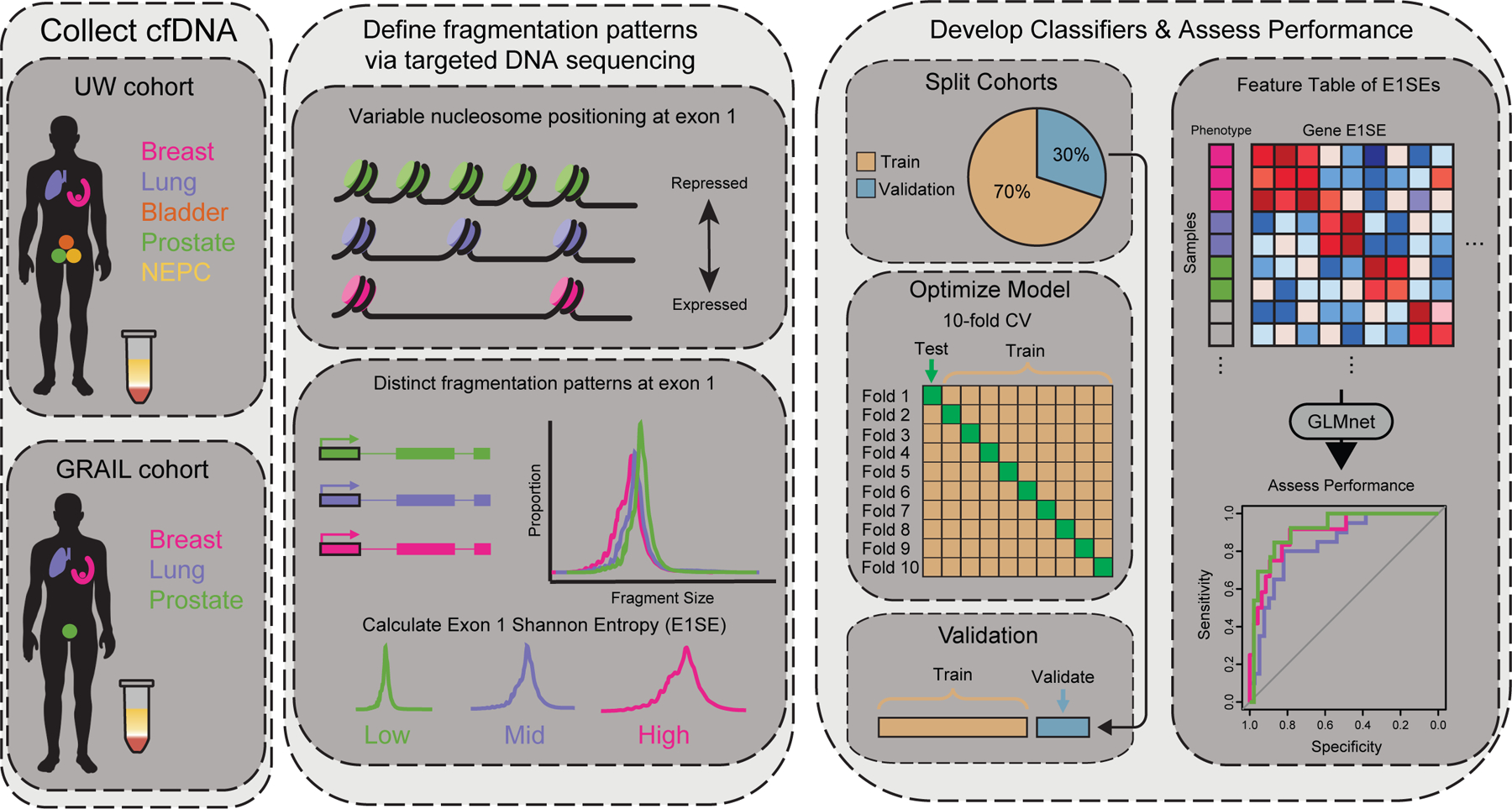
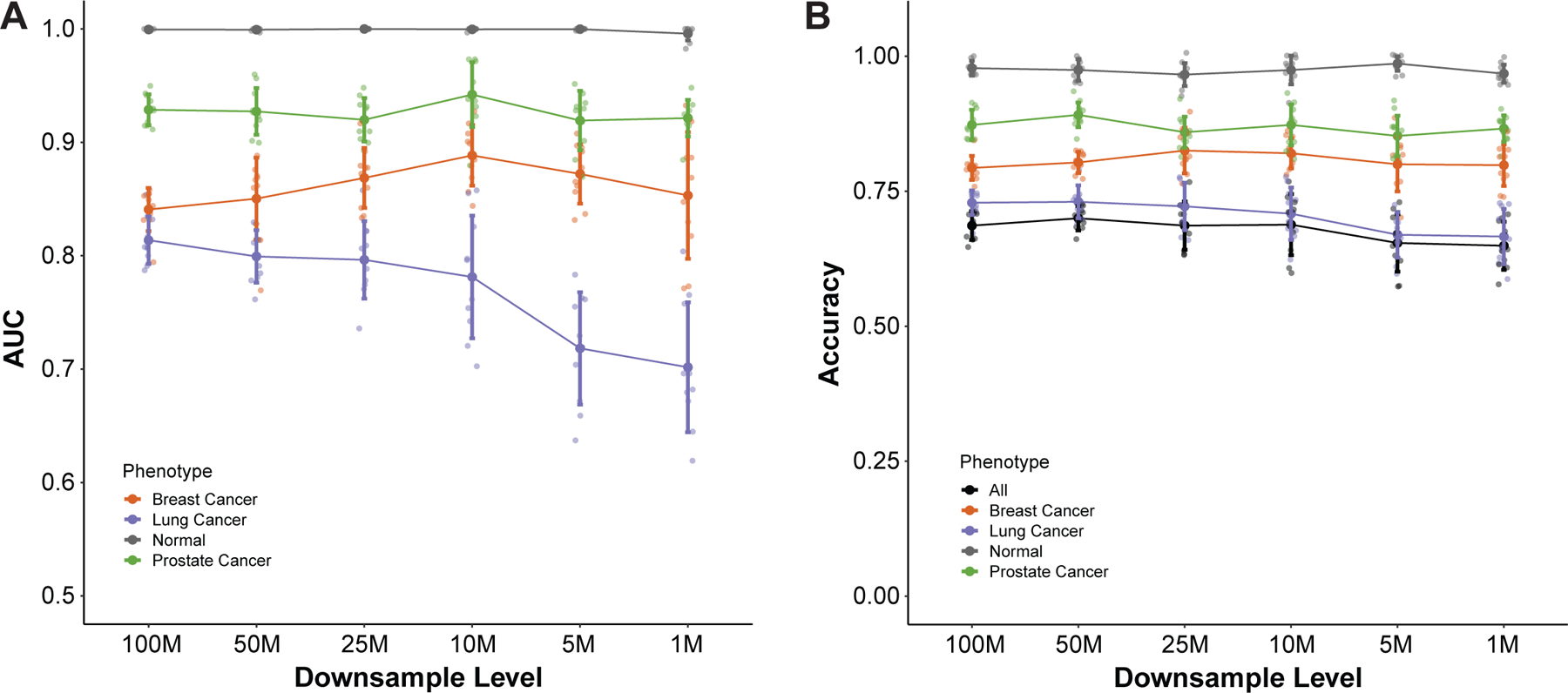
Patients and methods: We used machine learning models of fragmentation patterns at the first coding exon in standard targeted cancer gene cfDNA sequencing panels to distinguish between cancer and non-cancer patients, as well as the specific tumor type and subtype. We assessed this approach in two independent cohorts: a published cohort from GRAIL (breast, lung, and prostate cancers, non-cancer, n = 198) and an institutional cohort from the University of Wisconsin (UW; breast, lung, prostate, bladder cancers, n = 320). Each cohort was split 70%/30% into training and validation sets.
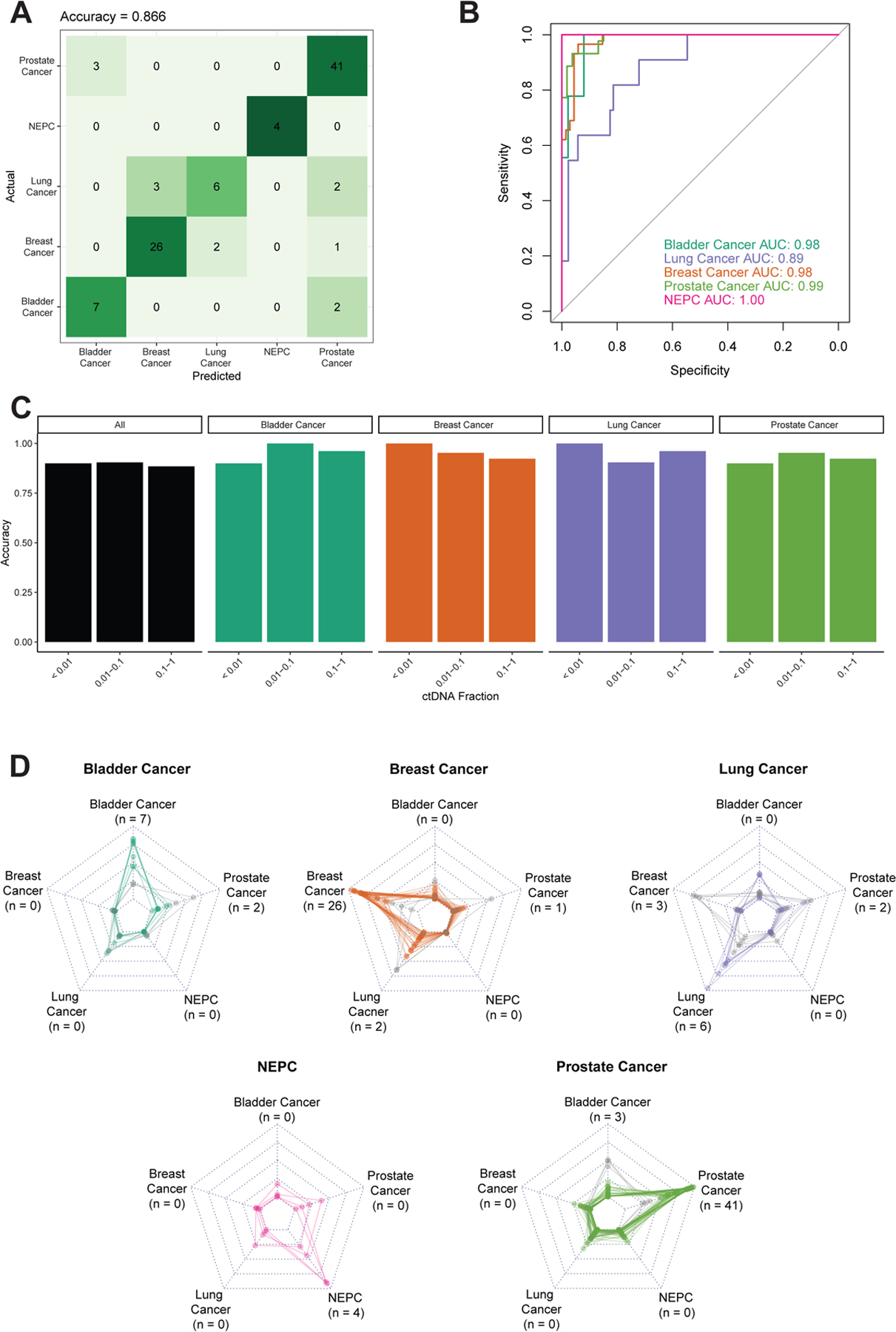
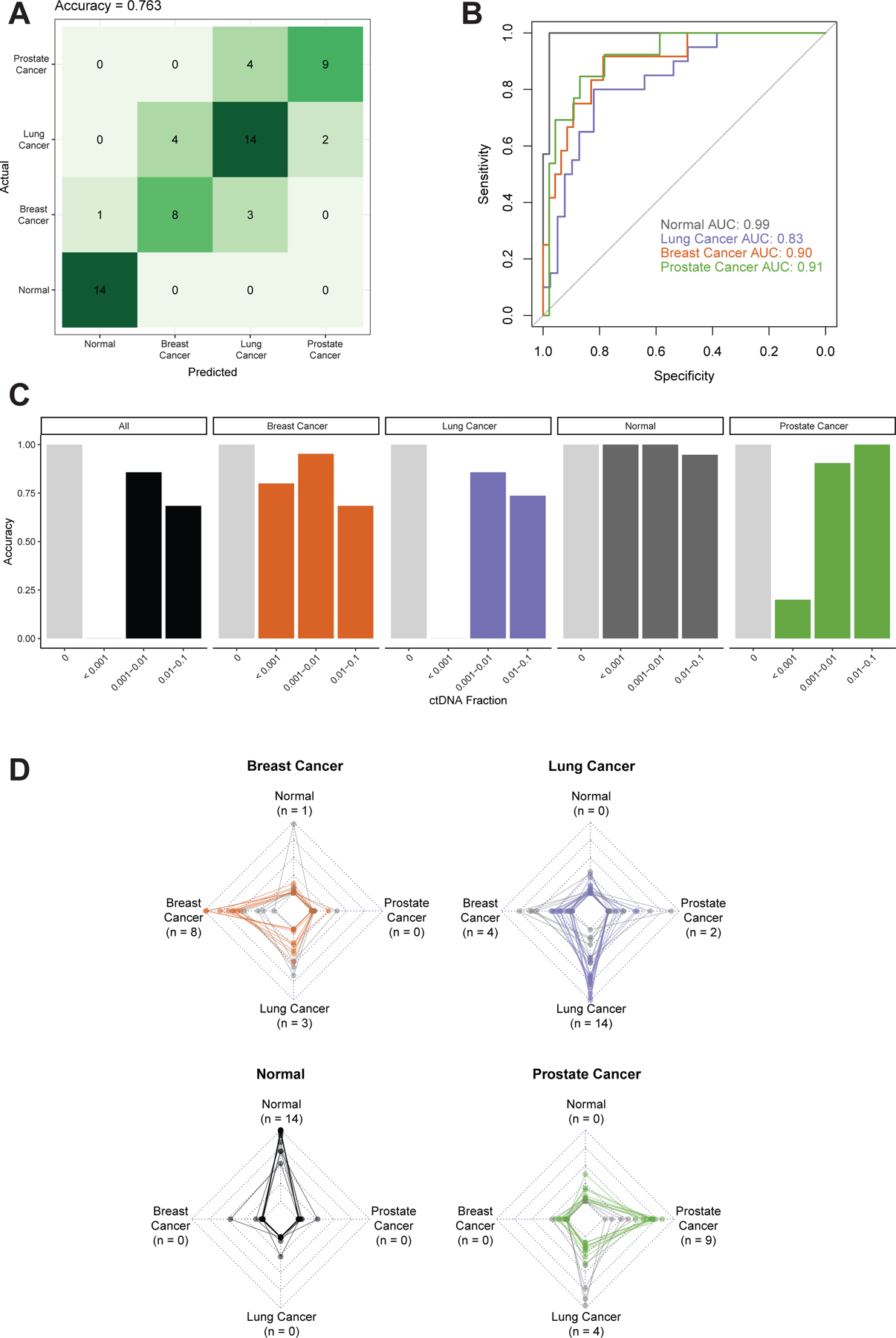
Results: In the UW cohort, training cross-validated accuracy was 82.1%, and accuracy in the independent validation cohort was 86.6% despite a median ctDNA fraction of only 0.06. In the GRAIL cohort, to assess how this approach performs in very low ctDNA fractions, training and independent validation were split based on ctDNA fraction. Training cross-validated accuracy was 80.6%, and accuracy in the independent validation cohort was 76.3%. In the validation cohort where the ctDNA fractions were all <0.05 and as low as 0.0003, the cancer versus non-cancer area under the curve was 0.99.
Conclusions: To our knowledge, this is the first study to demonstrate that sequencing from targeted cfDNA panels can be utilized to analyze fragmentation patterns to classify cancer types, dramatically expanding the potential capabilities of existing clinically used panels at minimal additional cost.
Keywords: cancer; cell-free DNA; fragmentomics.
Published by Elsevier Ltd.
Conflict of interest statement
Disclosure KTH has a family member who is an employee of Epic Systems. MLB has a family member who is an employee of Luminex. SGZ reports unrelated patents licensed to Veracyte, and that a family member is an employee of Artera and holds stock in Exact Sciences. KTH, SGZ, and the University of Wisconsin have filed a provisional patent on the work herein. SMD reports consulting relationships with BMS, Oncternal therapeutics, Janssen R&D/J&J and a grant from Pfizer/Astellas/Medivation (the grant was submitted to Medivation, ultimately funded by Astellas and then moved to Pfizer). FYF reports personal fees from Janssen Oncology, Bayer, PFS Genomics, Myovant Sciences, Roivant Sciences, Astellas Pharma, Foundation Medicine, Varian, Bristol Myers Squibb (BMS), Exact Sciences, BlueStar Genomics, Novartis, and Tempus; other support from Serimmune and Artera outside the submitted work. Integrated DNA Technologies (IDT, Coralville, IA) assisted in a pilot project to assess the performance characteristics of the UW panel before purchase, but played no role in this study. All other authors have declared no conflicts of interest. Data Sharing Raw sequencing data from the GRAIL dataset are available at the European Genome Archive (Dataset ID EGAD00001005302). Our institutional protocol does not allow unrestricted public access to the raw sequencing data. Therefore, data sharing requests must be submitted to the University of Wisconsin-Madison for approval. For samples from the two clinical trials (NCT03090165, NCT03725761), these trials are still ongoing, and data sharing requests must be submitted to the trial organizers.
Figures
References
-
- Yao W, Mei C, Nan X et al. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: A qualitative study. Gene 2016; 590 (1): 142–148. - PubMed
-
- Watanabe T, Takada S, Mizuta R. Cell-free DNA in blood circulation is generated by DNase1L3 and caspase-activated DNase. Biochem Biophys Res Commun 2019; 516 (3): 790–795. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
