

Soluble CD4 effectively prevents excessive TLR activation of resident macrophages in the onset of sepsis
- PMID: 37332010
- PMCID: PMC10277282
- DOI: 10.1038/s41392-023-01438-z
Soluble CD4 effectively prevents excessive TLR activation of resident macrophages in the onset of sepsis
Abstract
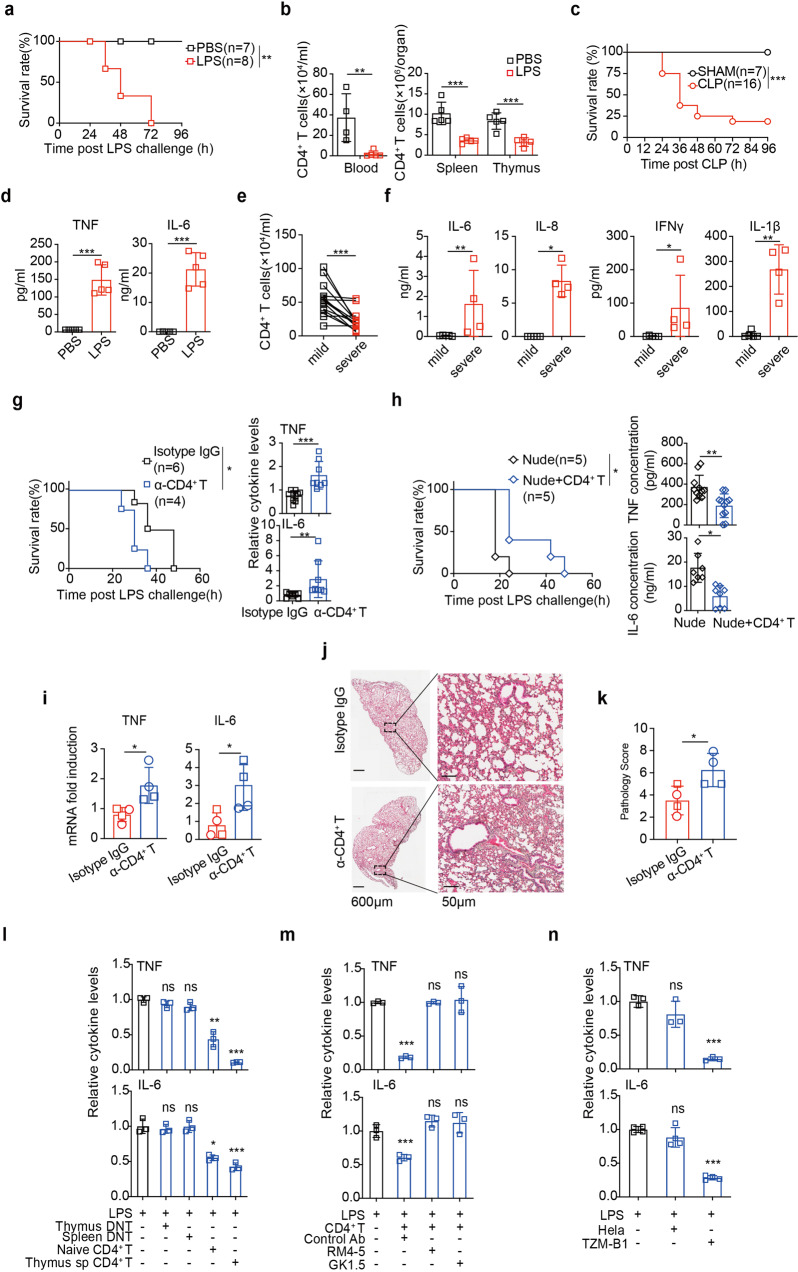
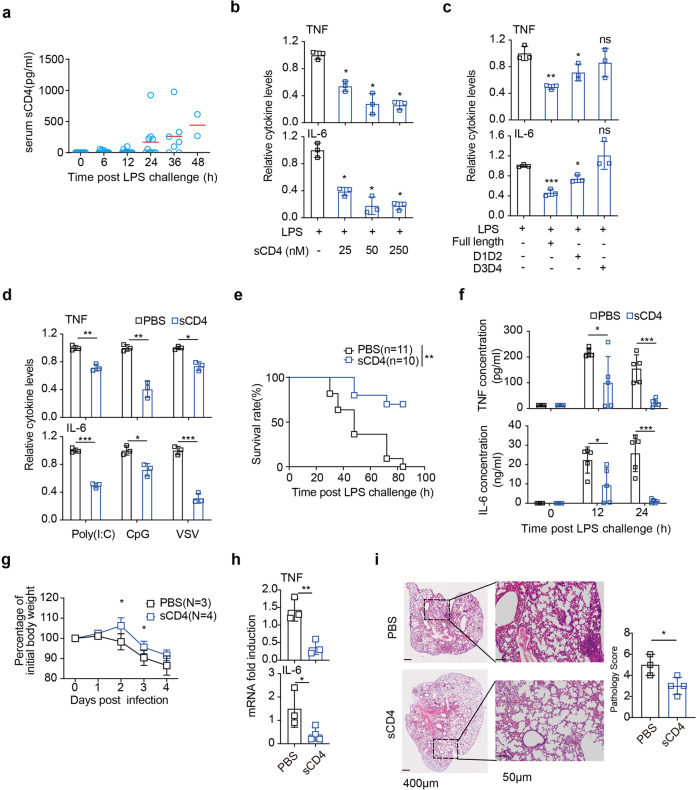
T lymphopenia, occurring in the early phase of sepsis in response to systemic inflammation, is commonly associated with morbidity and mortality of septic infections. We have previously shown that a sufficient number of T cells is required to constrain Toll-like receptors (TLRs) mediated hyperinflammation. However, the underlying mechanisms remains unsolved. Herein, we unveil that CD4+ T cells engage with MHC II of macrophages to downregulate TLR pro-inflammatory signaling. We show further that the direct contact between CD4 molecule of CD4+ T cells or the ectodomain of CD4 (soluble CD4, sCD4), and MHC II of resident macrophages is necessary and sufficient to prevent TLR4 overactivation in LPS and cecal ligation puncture (CLP) sepsis. sCD4 serum concentrations increase after the onset of LPS sepsis, suggesting its compensatory inhibitive effects on hyperinflammation. sCD4 engagement enables the cytoplasmic domain of MHC II to recruit and activate STING and SHP2, which inhibits IRAK1/Erk and TRAF6/NF-κB activation required for TLR4 inflammation. Furthermore, sCD4 subverts pro-inflammatory plasma membrane anchorage of TLR4 by disruption of MHC II-TLR4 raft domains that promotes MHC II endocytosis. Finally, sCD4/MHCII reversal signaling specifically interferes with TLR4 but not TNFR hyperinflammation, and independent of the inhibitive signaling of CD40 ligand of CD4+ cells on macrophages. Therefore, a sufficient amount of soluble CD4 protein can prevent excessive inflammatory activation of macrophages via alternation of MHC II-TLR signaling complex, that might benefit for a new paradigm of preventive treatment of sepsis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
