

Anderson-Fabry disease cardiomyopathy: an update on epidemiology, diagnostic approach, management and monitoring strategies
- PMID: 37332587
- PMCID: PMC10272370
- DOI: 10.3389/fcvm.2023.1152568
Anderson-Fabry disease cardiomyopathy: an update on epidemiology, diagnostic approach, management and monitoring strategies
Abstract
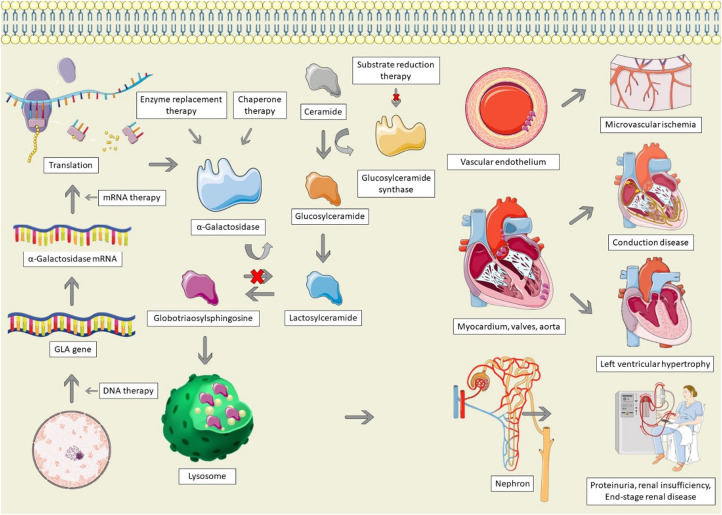
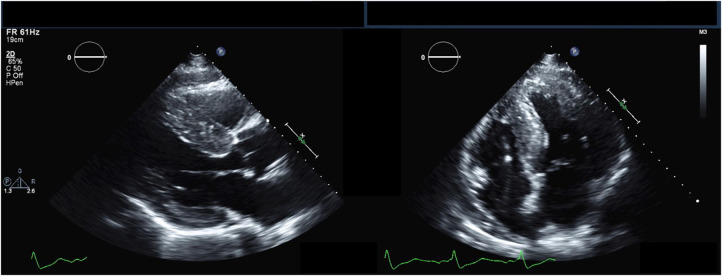
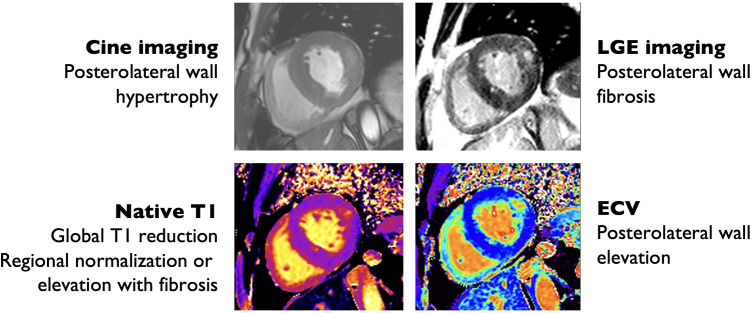
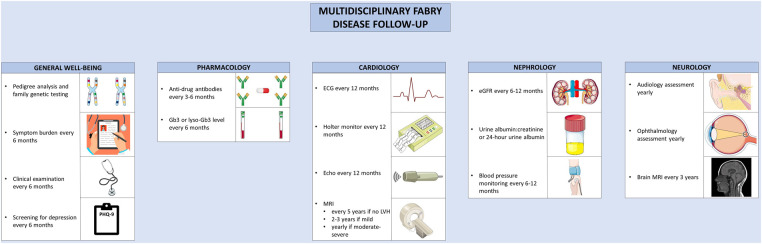
Anderson-Fabry disease (AFD) is an X-linked lysosomal storage disorder caused by deficient activity of the enzyme alpha-galactosidase. While AFD is recognized as a progressive multi-system disorder, infiltrative cardiomyopathy causing a number of cardiovascular manifestations is recognized as an important complication of this disease. AFD affects both men and women, although the clinical presentation typically varies by sex, with men presenting at a younger age with more neurologic and renal phenotype and women developing a later onset variant with more cardiovascular manifestations. AFD is an important cause of increased myocardial wall thickness, and advances in imaging, in particular cardiac magnetic resonance imaging and T1 mapping techniques, have improved the ability to identify this disease non-invasively. Diagnosis is confirmed by the presence of low alpha-galactosidase activity and identification of a mutation in the GLA gene. Enzyme replacement therapy remains the mainstay of disease modifying therapy, with two formulations currently approved. In addition, newer treatments such as oral chaperone therapy are now available for select patients, with a number of other investigational therapies in development. The availability of these therapies has significantly improved outcomes for AFD patients. Improved survival and the availability of multiple agents has presented new clinical dilemmas regarding disease monitoring and surveillance using clinical, imaging and laboratory biomarkers, in addition to improved approaches to managing cardiovascular risk factors and AFD complications. This review will provide an update on clinical recognition and diagnostic approaches including differentiation from other causes of increased ventricular wall thickness, in addition to modern strategies for management and follow-up.
Keywords: anderson-Fabry disease; cardiac imaging; enzyme replacement therapy; heart failure; management.
© 2023 Averbuch, White and Fine.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Anderson-Fabry cardiomyopathy: prevalence, pathophysiology, diagnosis and treatment.Heart Fail Rev. 2015 Mar;20(2):179-91. doi: 10.1007/s10741-014-9452-9. Heart Fail Rev. 2015. PMID: 25030479 Review.
-
Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson-Fabry Disease.Int J Mol Sci. 2024 Apr 12;25(8):4299. doi: 10.3390/ijms25084299. Int J Mol Sci. 2024. PMID: 38673884 Free PMC article.
-
Advanced Anderson-Fabry disease presenting with left ventricular apical aneurysm and ventricular tachycardia.World J Clin Cases. 2015 Jun 16;3(6):519-24. doi: 10.12998/wjcc.v3.i6.519. World J Clin Cases. 2015. PMID: 26090373 Free PMC article.
-
Prevalence of lymphedema among Anderson-Fabry disease patients: A report from the Fabry registry.Mol Genet Metab. 2023 Apr;138(4):107538. doi: 10.1016/j.ymgme.2023.107538. Epub 2023 Feb 8. Mol Genet Metab. 2023. PMID: 36812723
-
The role of cardiovascular multimodality imaging in the evaluation of Anderson-Fabry disease: from early diagnosis to therapy monitoring.Eur Heart J Cardiovasc Imaging. 2025 Apr 30;26(5):814-829. doi: 10.1093/ehjci/jeaf038. Eur Heart J Cardiovasc Imaging. 2025. PMID: 39903606
Cited by
-
From thick walls to clear answers: approaches to diagnosing hypertrophic cardiomyopathy and its mimics.Eur Heart J Suppl. 2025 Feb 19;27(Suppl 1):i40-i46. doi: 10.1093/eurheartjsupp/suae099. eCollection 2025 Feb. Eur Heart J Suppl. 2025. PMID: 39980777 Free PMC article.
-
Enzyme Replacement and Immunosuppression in Heart Transplant Recipients with Fabry Cardiomyopathy: A 7-Year Case Study.Am J Case Rep. 2025 Mar 17;26:e945873. doi: 10.12659/AJCR.945873. Am J Case Rep. 2025. PMID: 40095984 Free PMC article.
-
Hydroxychloroquine Cardiotoxicity and Sarcomeric Hypertrophic Cardiomyopathy.JACC Case Rep. 2025 Jun 11;30(14):103683. doi: 10.1016/j.jaccas.2025.103683. JACC Case Rep. 2025. PMID: 40514126 Free PMC article.
-
Incremental diagnostic and prognostic utility of left atrial deformation in heart failure using speckle tracking echocardiography.Heart Fail Rev. 2024 May;29(3):713-727. doi: 10.1007/s10741-024-10392-z. Epub 2024 Mar 11. Heart Fail Rev. 2024. PMID: 38466374 Review.
-
Cardiopulmonary determinants of reduced exercise tolerance in Fabry disease.Front Cardiovasc Med. 2024 May 2;11:1396996. doi: 10.3389/fcvm.2024.1396996. eCollection 2024. Front Cardiovasc Med. 2024. PMID: 38756750 Free PMC article. Review.
References
-
- Bartolotta C, Filogamo M, Colomba P, Zizzo C, Albeggiani G, Scalia S, et al. FP907History of Anderson—Fabry disease. Nephrol Dial Transplant. (2015) 30(suppl_3):iii379. 10.1093/ndt/gfv186.08 - DOI
-
- Germain DP, Brand E, Cecchi F, Kempf J, Laney DA, Linhart A, et al. The phenotypic characteristics of the p.N215S Fabry disease genotype in male and female patients: a multi-center Fabry Registry study. Mol Genet Metab. (2017) 120(1–2):S51–2. 10.1002/mgg3.389 - DOI
Publication types
LinkOut - more resources
Full Text Sources