

This is a preprint.
Calling Cards: a customizable platform to longitudinally record protein-DNA interactions over time in cells and tissues
- PMID: 37333130
- PMCID: PMC10274760
- DOI: 10.1101/2023.06.07.544098
Calling Cards: a customizable platform to longitudinally record protein-DNA interactions over time in cells and tissues
Update in
-
Calling Cards: A Customizable Platform to Longitudinally Record Protein-DNA Interactions Over Time in Cells and Tissues.Curr Protoc. 2023 Sep;3(9):e883. doi: 10.1002/cpz1.883. Curr Protoc. 2023. PMID: 37755132 Free PMC article.
Abstract
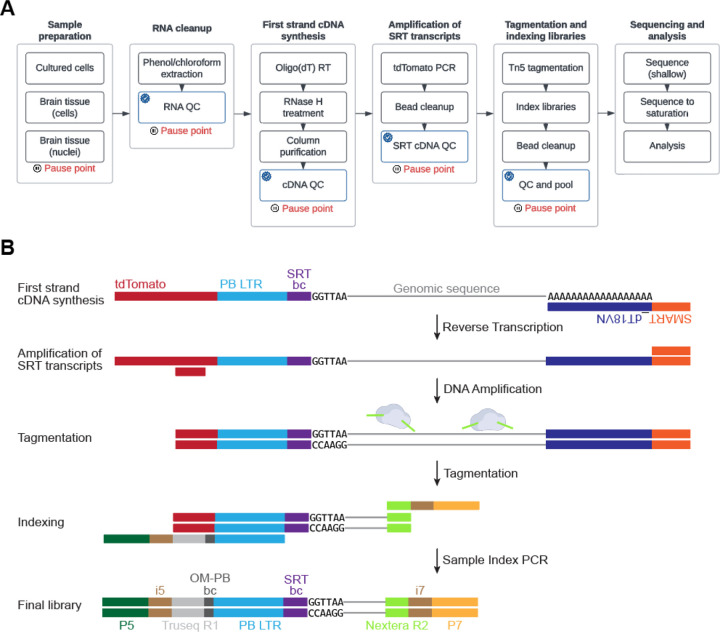
Calling Cards is a platform technology to record a cumulative history of transient protein-DNA interactions in the genome of genetically targeted cell types. The record of these interactions is recovered by next generation sequencing. Compared to other genomic assays, whose readout provides a snapshot at the time of harvest, Calling Cards enables correlation of historical molecular states to eventual outcomes or phenotypes. To achieve this, Calling Cards uses the piggyBac transposase to insert self-reporting transposon (SRT) "Calling Cards" into the genome, leaving permanent marks at interaction sites. Calling Cards can be deployed in a variety of in vitro and in vivo biological systems to study gene regulatory networks involved in development, aging, and disease. Out of the box, it assesses enhancer usage but can be adapted to profile specific transcription factor binding with custom transcription factor (TF)-piggyBac fusion proteins. The Calling Cards workflow has five main stages: delivery of Calling Card reagents, sample preparation, library preparation, sequencing, and data analysis. Here, we first present a comprehensive guide for experimental design, reagent selection, and optional customization of the platform to study additional TFs. Then, we provide an updated protocol for the five steps, using reagents that improve throughput and decrease costs, including an overview of a newly deployed computational pipeline. This protocol is designed for users with basic molecular biology experience to process samples into sequencing libraries in 1-2 days. Familiarity with bioinformatic analysis and command line tools is required to set up the pipeline in a high-performance computing environment and to conduct downstream analyses. Basic Protocol 1: Preparation and delivery of Calling Cards reagentsBasic Protocol 2: Sample preparationBasic Protocol 3: Sequencing library preparationBasic Protocol 4: Library pooling and sequencingBasic Protocol 5: Data analysis.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
RDM and AM are listed as inventors on US patent US20200181626A1.
Figures










References
-
- Cammack A. J., Moudgil A., Chen J., Vasek M. J., Shabsovich M., McCullough K., Yen A., Lagunas T., Maloney S. E., He J., et al. 2020. A viral toolkit for recording transcription factor–DNA interactions in live mouse tissues. Proceedings of the National Academy of Sciences 117:10003–10014. - PMC - PubMed
-
- Challis R. C., Ravindra Kumar S., Chan K. Y., Challis C., Beadle K., Jang M. J., Kim H. M., Rajendran P. S., Tompkins J. D., Shivkumar K., et al. 2019. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nature Protocols 14:379–414. - PubMed
INTERNET RESOURCES
-
- https://nf-co.re/callingcards: This is the official nf-core community page that hosts the bioinformatics pipeline. Complete documentation and release notes can be found here.
-
- https://github.com/nf-core/callingcards/issues: Found a bug? Have a feature request? We welcome any submissions big or small through github.
-
- https://nfcore.slack.com/channels/callingcards: This is the official slack channel that is monitored by the developers and authors. Feel free to drop in to ask questions or just say ‘hi’!
-
- https://www.addgene.org/kits/mitra-barcoded-transposon/: This is a link to an Addgene plasmid kit that contains individual barcoded self-reporting transposons. These can be grown up and pooled into one large pool or multiple subpools.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous