

Multicellular communities are perturbed in the aging human brain and Alzheimer's disease
- PMID: 37336975
- PMCID: PMC10789499
- DOI: 10.1038/s41593-023-01356-x
Multicellular communities are perturbed in the aging human brain and Alzheimer's disease
Abstract
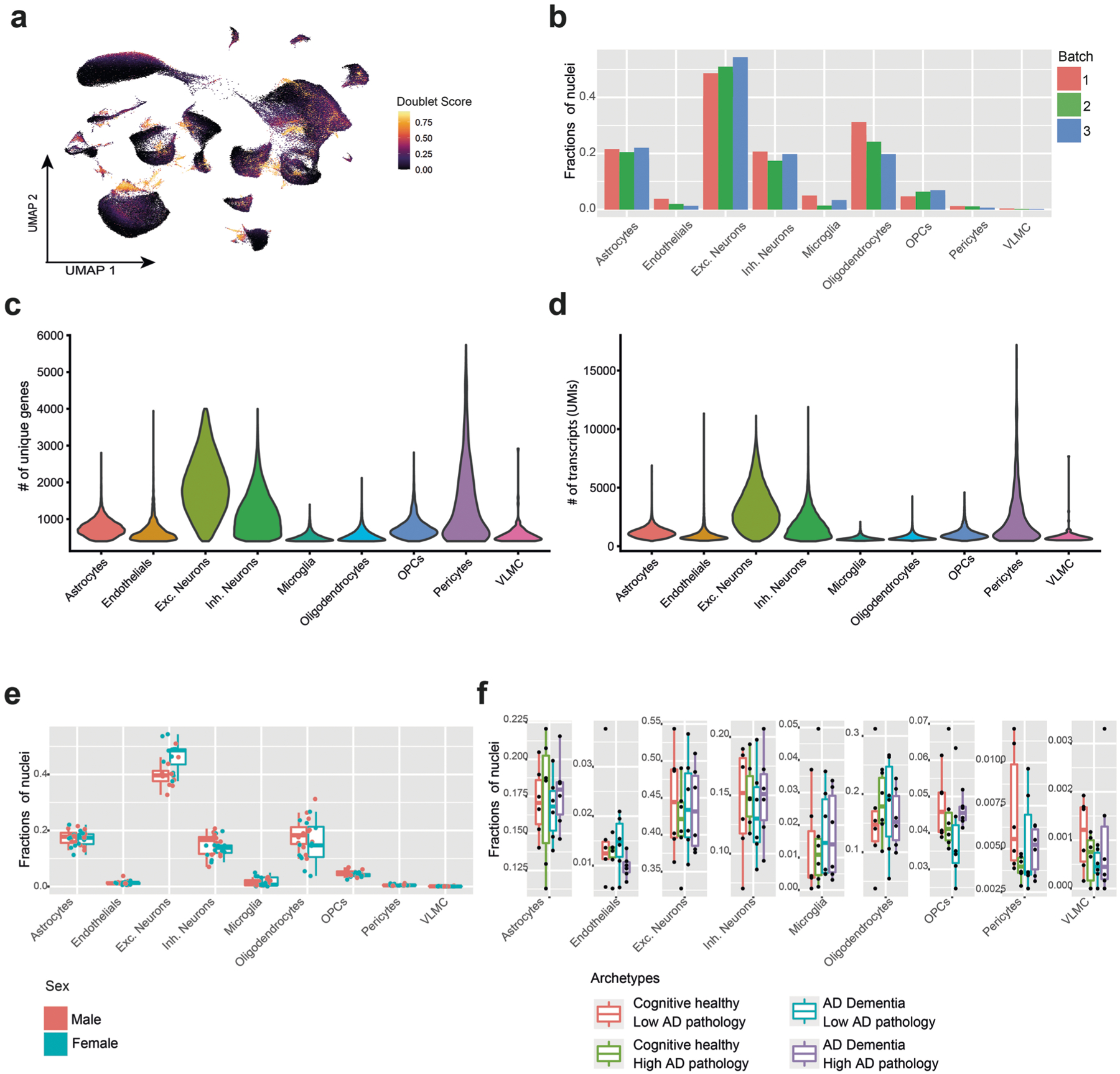
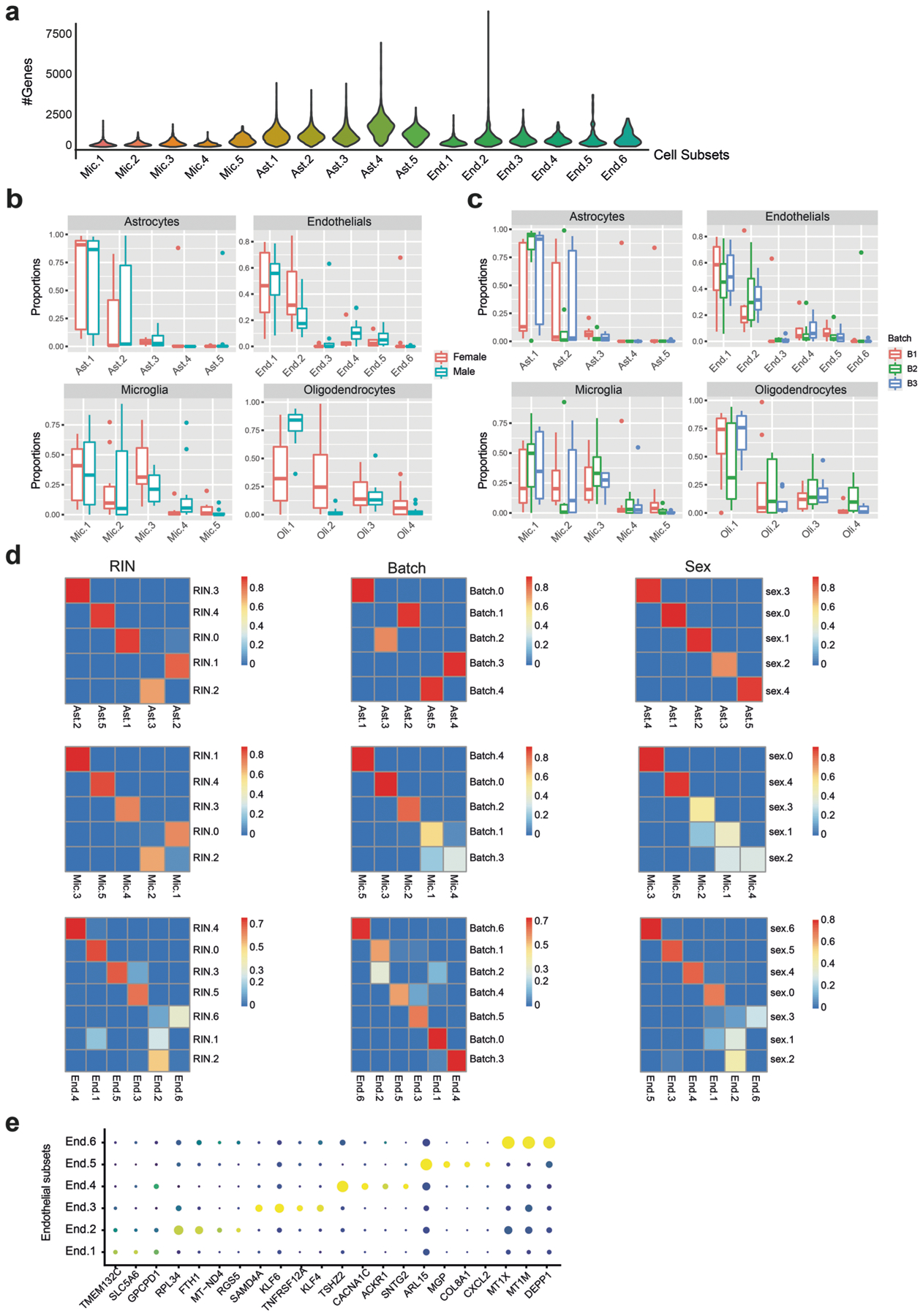
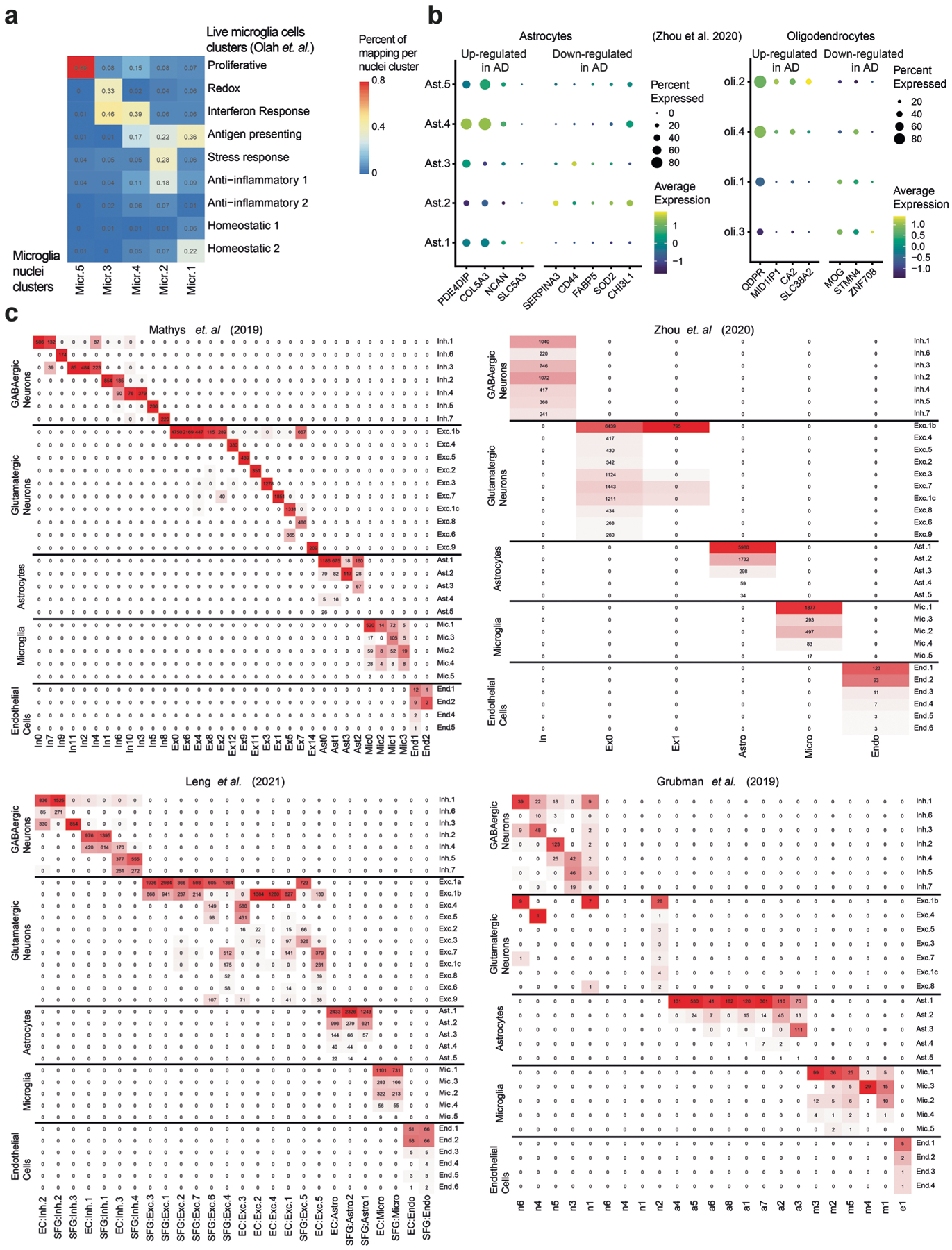
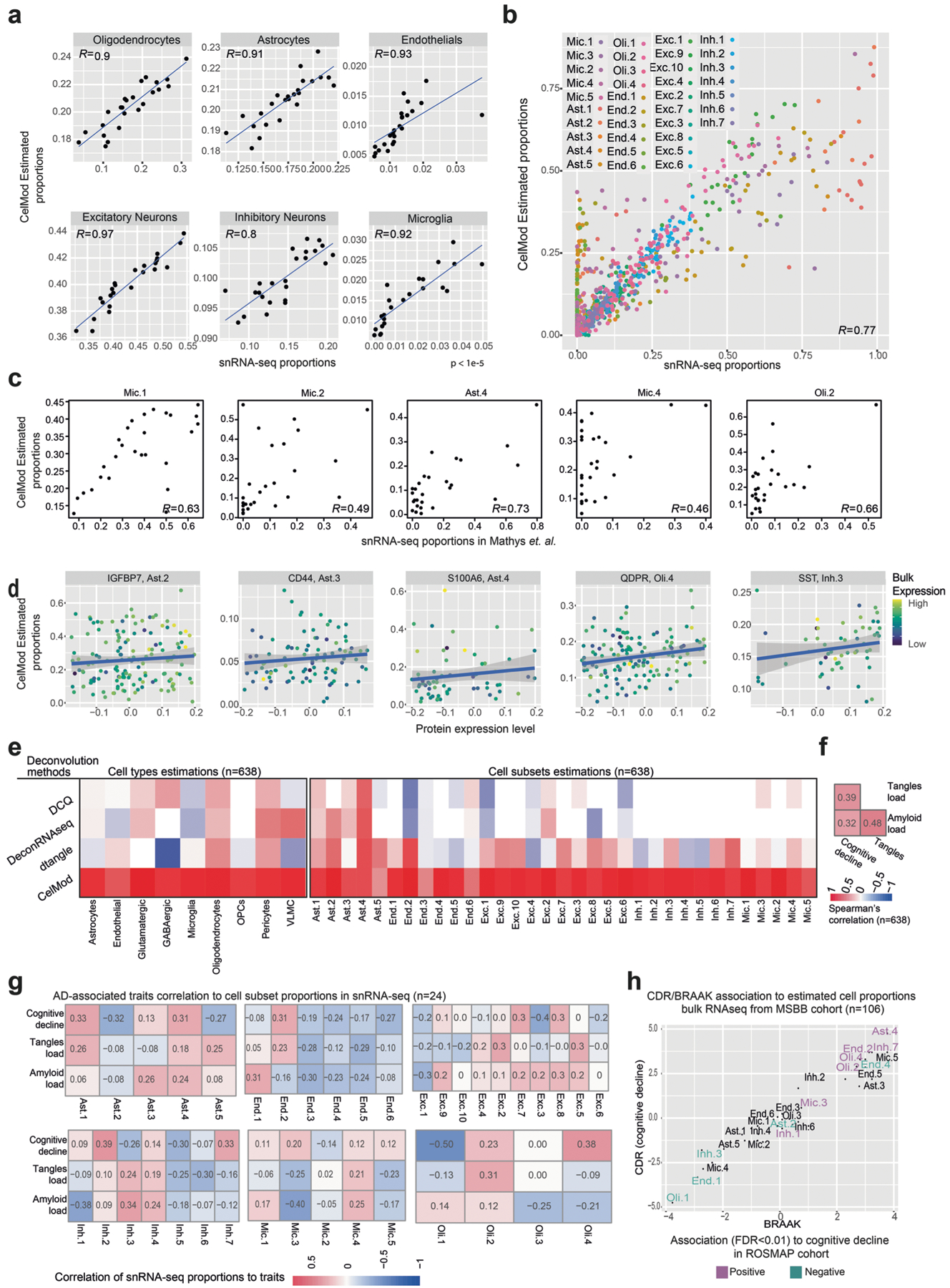
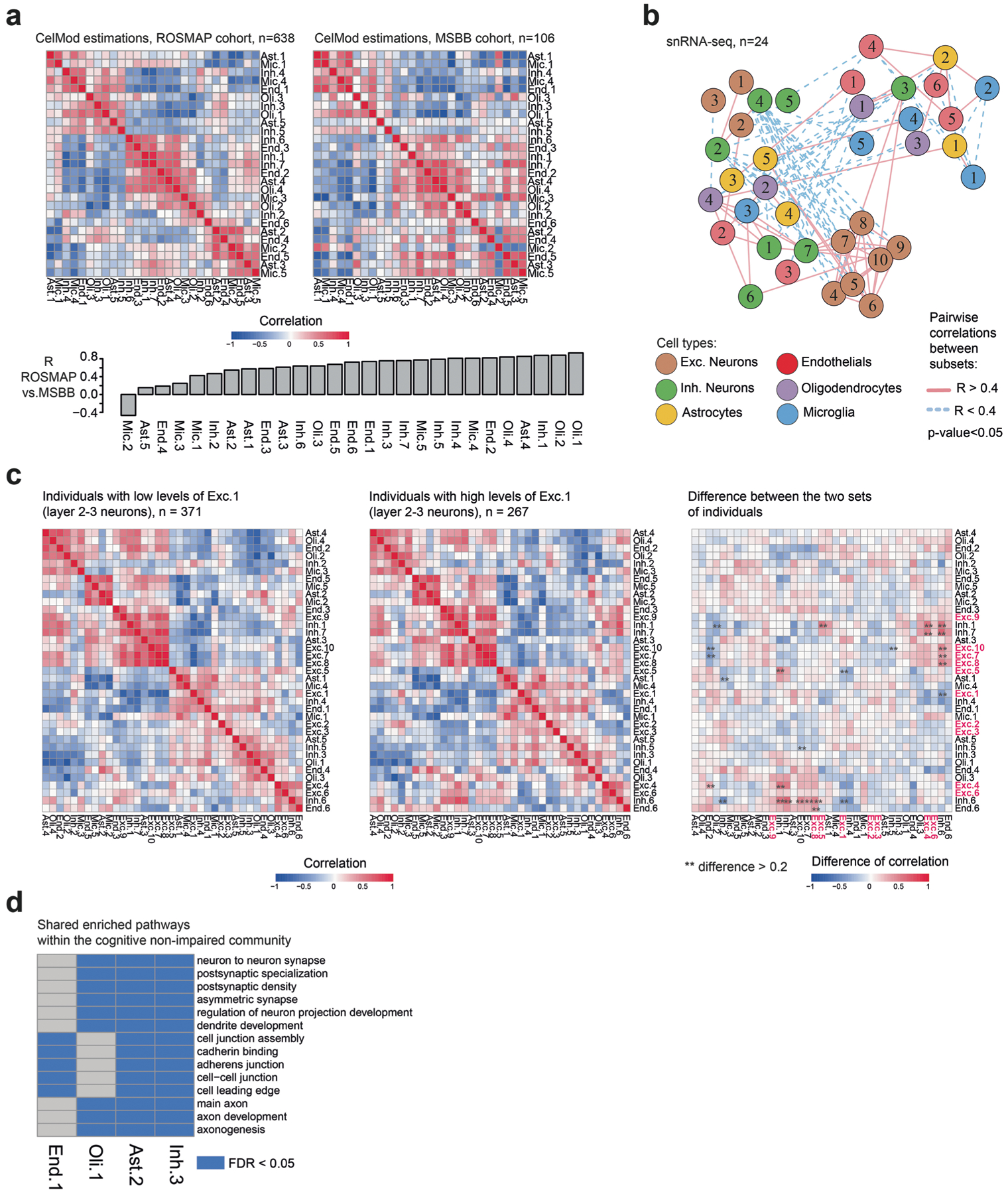
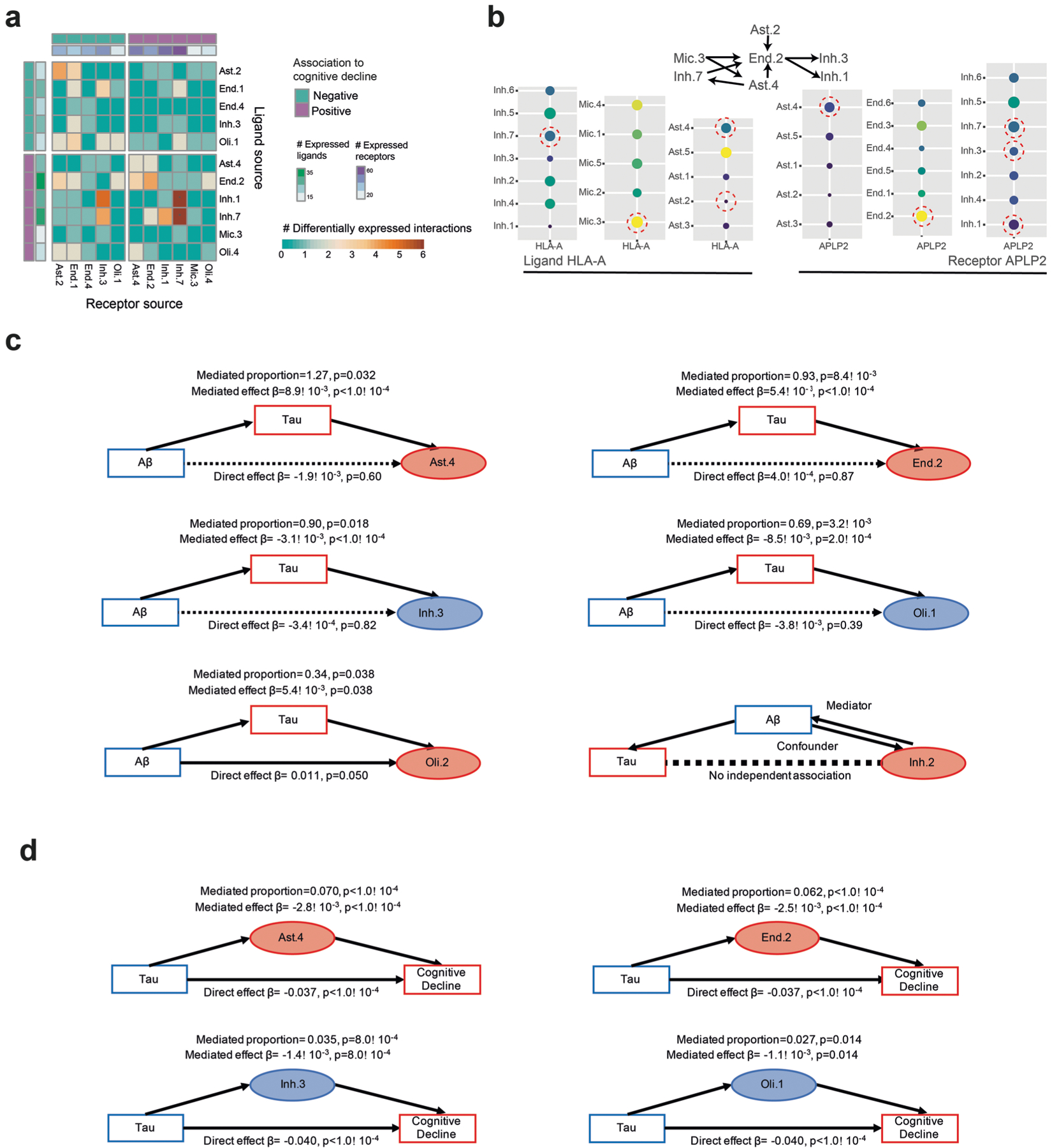
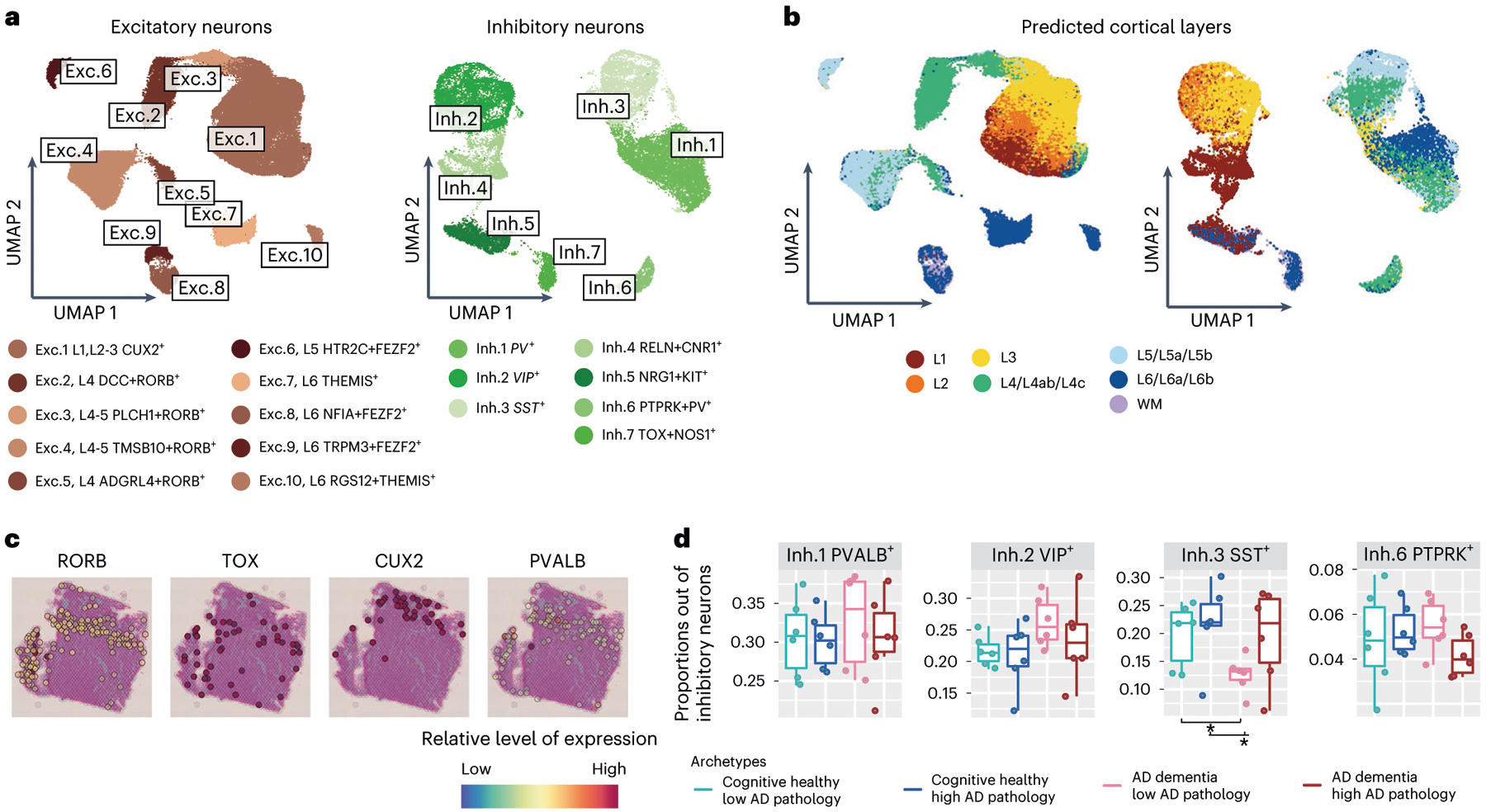
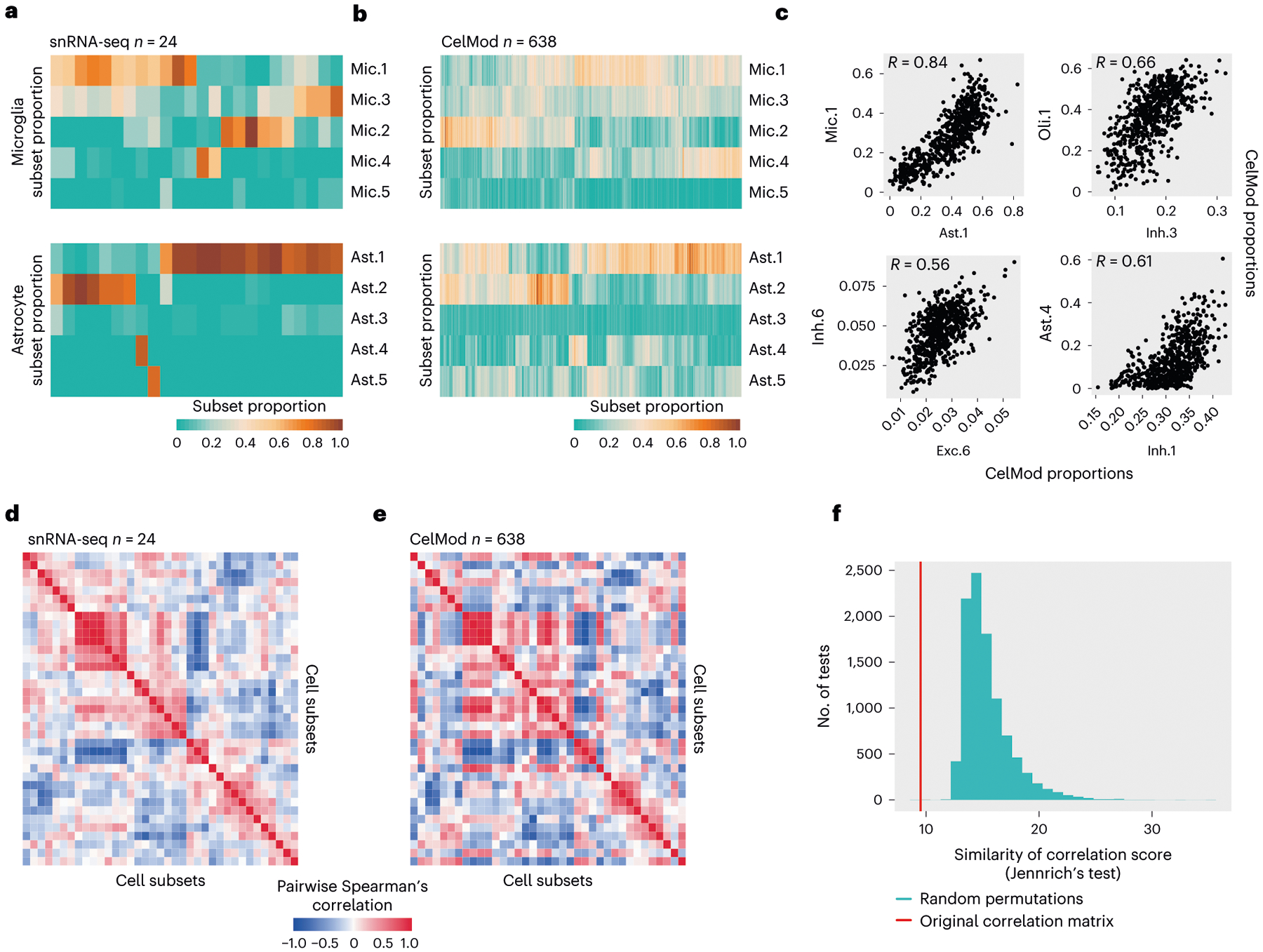
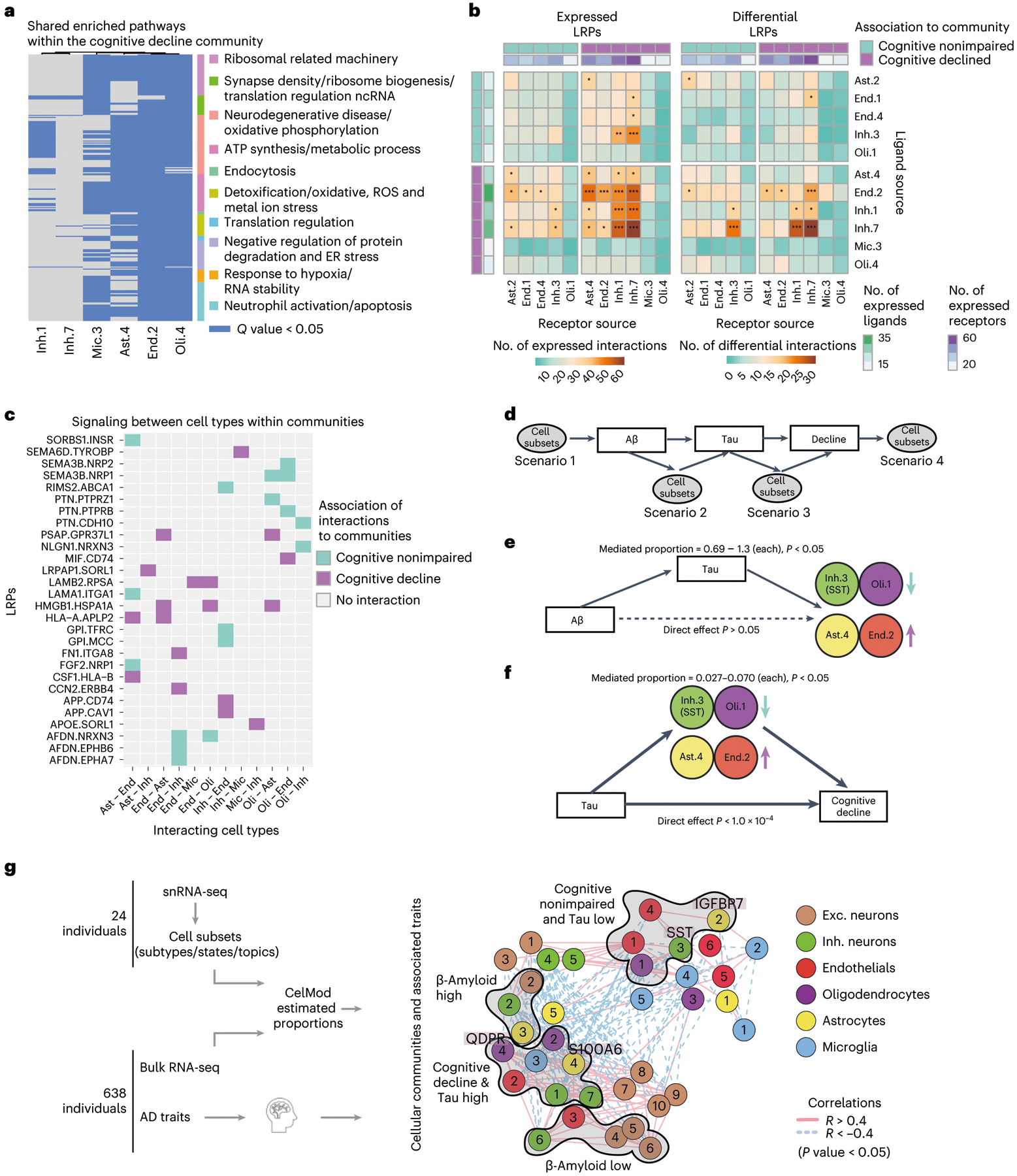
The role of different cell types and their interactions in Alzheimer's disease (AD) is a complex and open question. Here, we pursued this question by assembling a high-resolution cellular map of the aging frontal cortex using single-nucleus RNA sequencing of 24 individuals with a range of clinicopathologic characteristics. We used this map to infer the neocortical cellular architecture of 638 individuals profiled by bulk RNA sequencing, providing the sample size necessary for identifying statistically robust associations. We uncovered diverse cell populations associated with AD, including a somatostatin inhibitory neuronal subtype and oligodendroglial states. We further identified a network of multicellular communities, each composed of coordinated subpopulations of neuronal, glial and endothelial cells, and we found that two of these communities are altered in AD. Finally, we used mediation analyses to prioritize cellular changes that might contribute to cognitive decline. Thus, our deconstruction of the aging neocortex provides a roadmap for evaluating the cellular microenvironments underlying AD and dementia.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until 31 August 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. From 1 August 2020, A.R. is an employee of Genentech, a member of the Roche Group. The remaining authors declare no competing interests.
Figures






References
-
- Grubman A et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
