

Genetic characterization of non-5q proximal spinal muscular atrophy in a French cohort: the place of whole exome sequencing
- PMID: 37337091
- PMCID: PMC10772122
- DOI: 10.1038/s41431-023-01407-8
Genetic characterization of non-5q proximal spinal muscular atrophy in a French cohort: the place of whole exome sequencing
Abstract
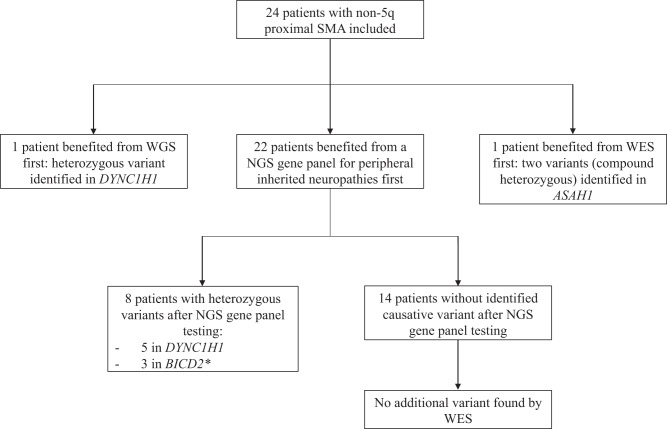
Proximal spinal muscular atrophy (SMA) is defined by a degeneration of the anterior horn cells resulting in muscle weakness predominantly in the proximal lower limbs. While most patients carry a biallelic deletion in the SMN1 gene (localized in chromosome 5q), little is known regarding patients without SMN1-mutation, and a genetic diagnosis is not always possible. Here, we report a cohort of 24 French patients with non-5q proximal SMA from five neuromuscular centers who all, except two, had next-generation sequencing (NGS) gene panel, followed by whole exome sequencing (WES) if gene panel showed a negative result. The two remaining patients benefited directly from WES or whole genome sequencing (WGS). A total of ten patients with causative variants were identified, nine of whom were index cases (9/23 families = 39%). Eight variants were identified by gene panel: five variants in DYNC1H1, and three in BICD2. Compound heterozygous causative variants in ASAH1 were identified directly by WES, and one variant in DYNC1H1 was identified directly by WGS. No causative variant was found using WES in patients with a previous panel with negative results (14 cases). We thus recommend using primarily NGS panels in patients with non-5q-SMA and using WES, especially when several members of the same family are affected and/or when trio analyses are possible, or WGS as second-line testing if available.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Arnold ES, Fischbeck KH. Spinal muscular atrophy. In: Handbook of Clinical Neurology. Elsevier; 2018. p. 591–601. https://linkinghub.elsevier.com/retrieve/pii/B9780444640765000387. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
