

Epichaperome inhibition targets TP53-mutant AML and AML stem/progenitor cells
- PMID: 37339579
- PMCID: PMC10656725
- DOI: 10.1182/blood.2022019047
Epichaperome inhibition targets TP53-mutant AML and AML stem/progenitor cells
Abstract
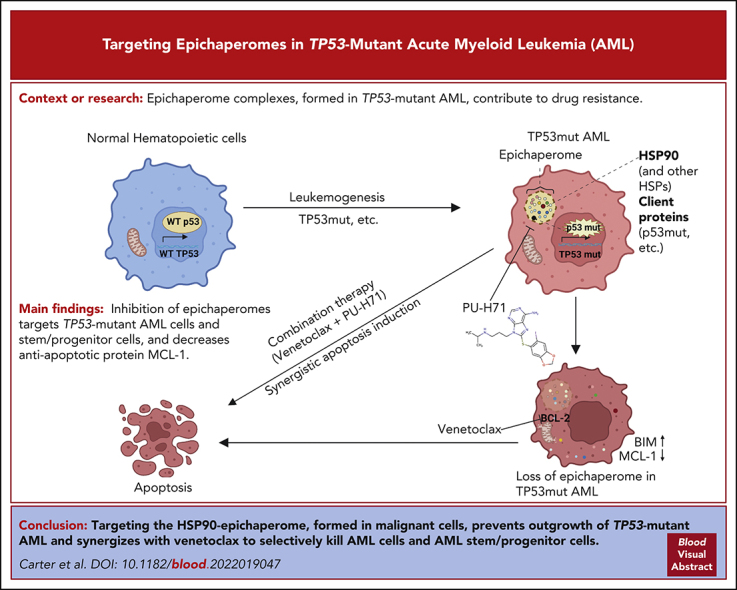
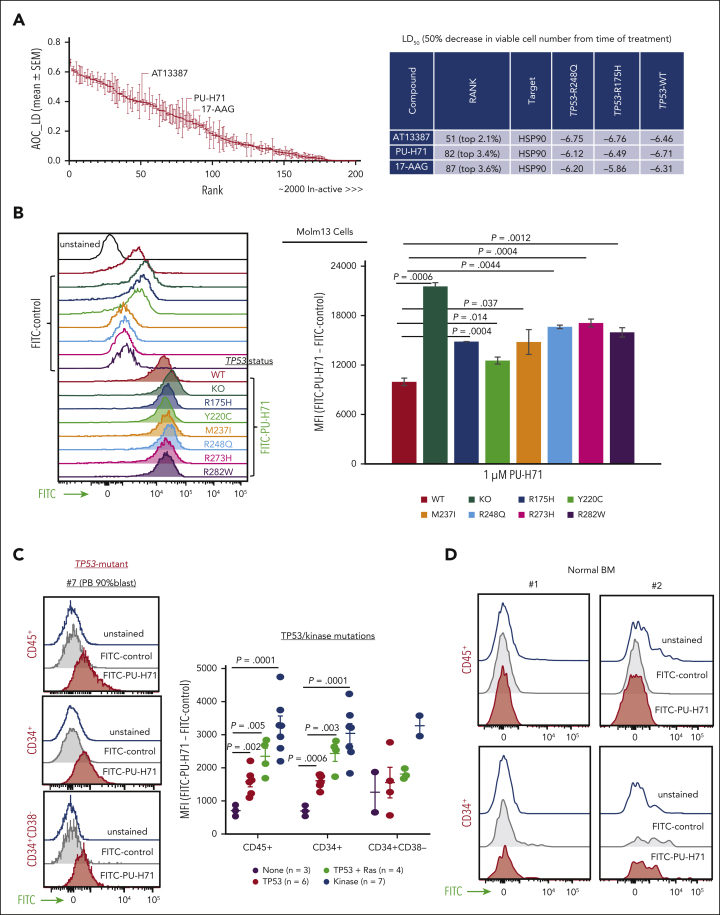
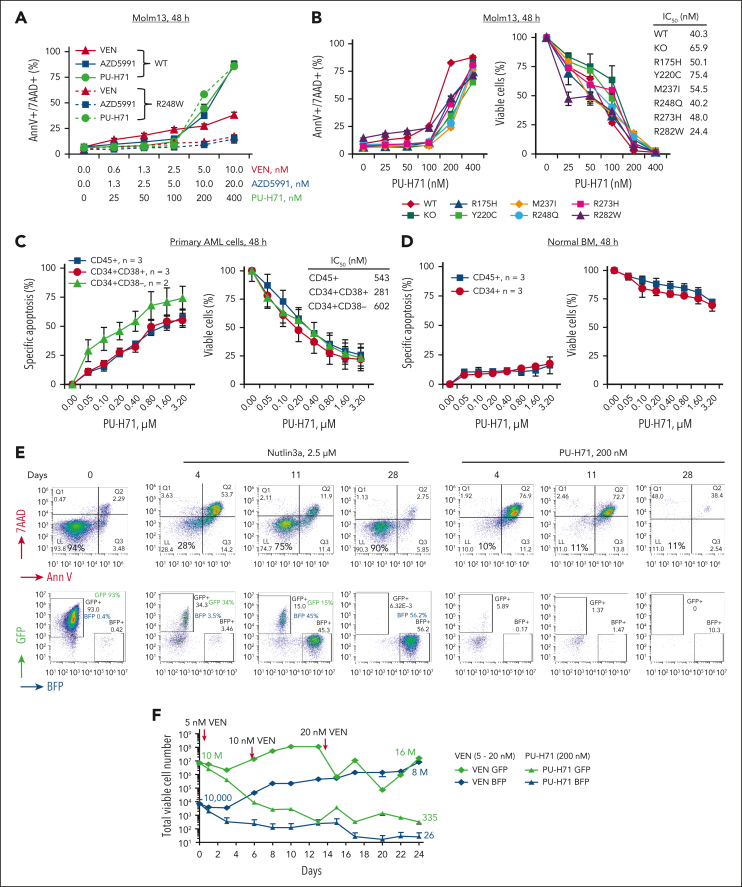
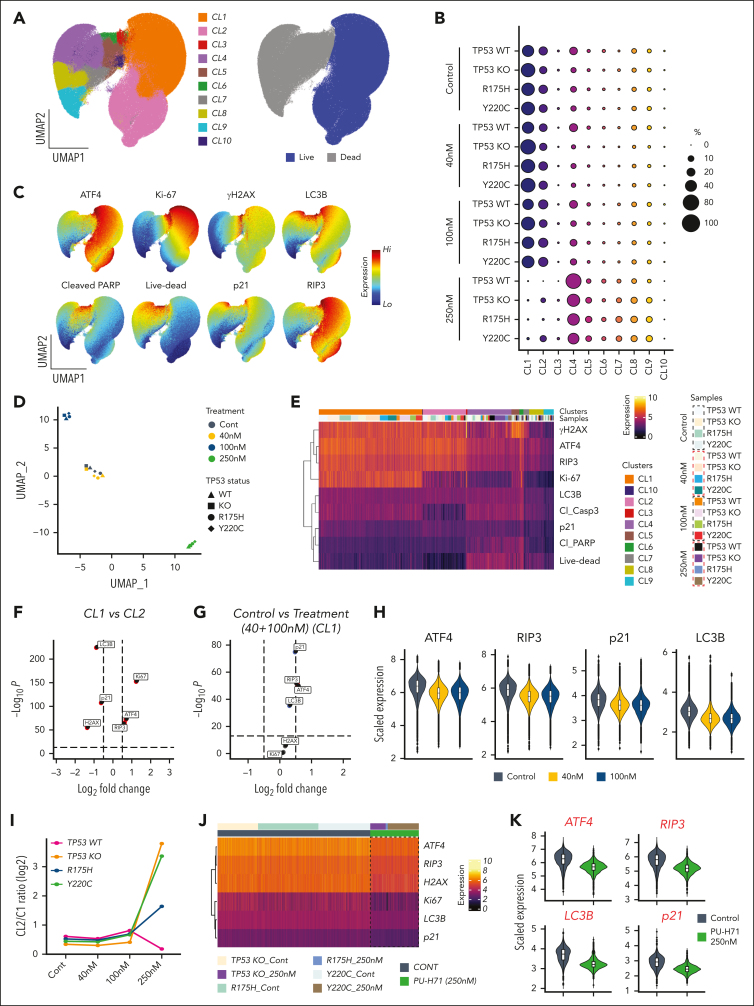
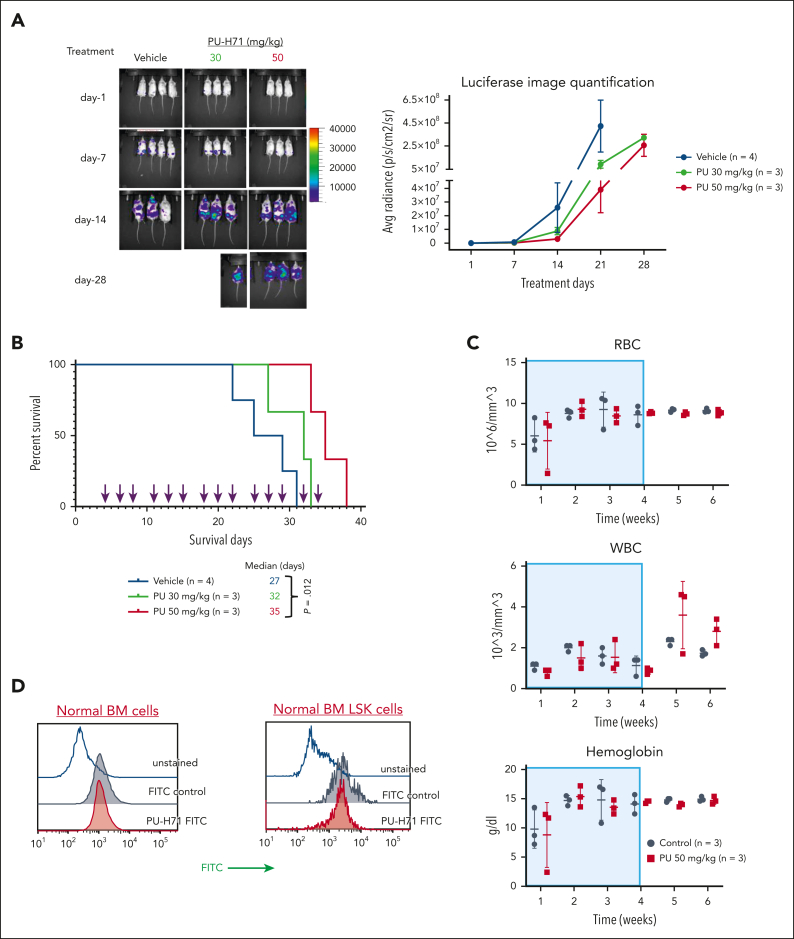
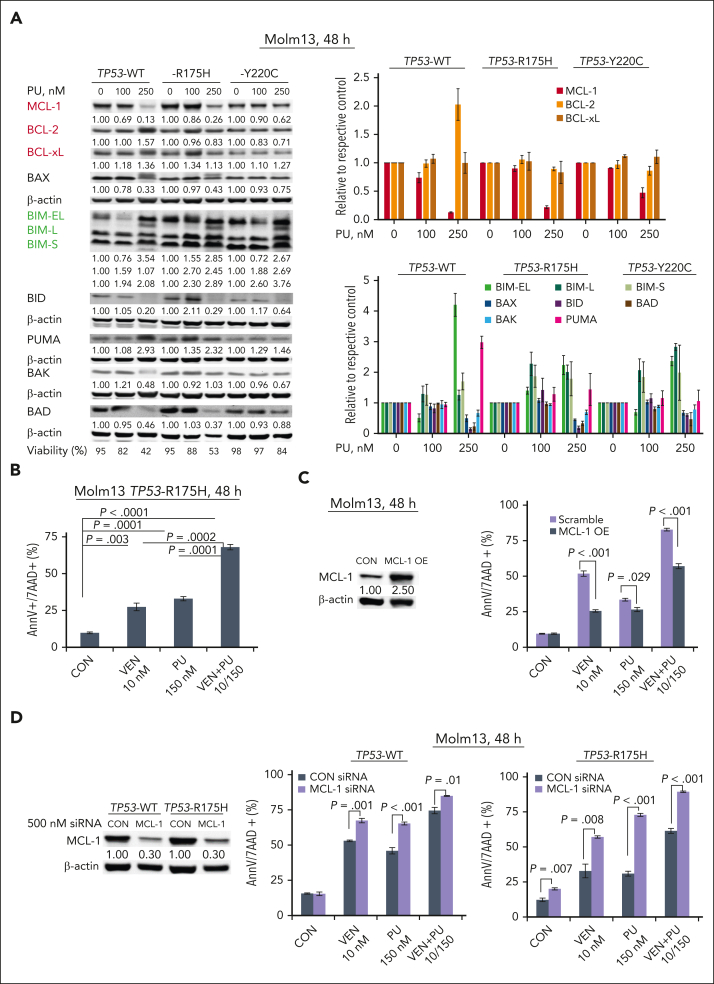
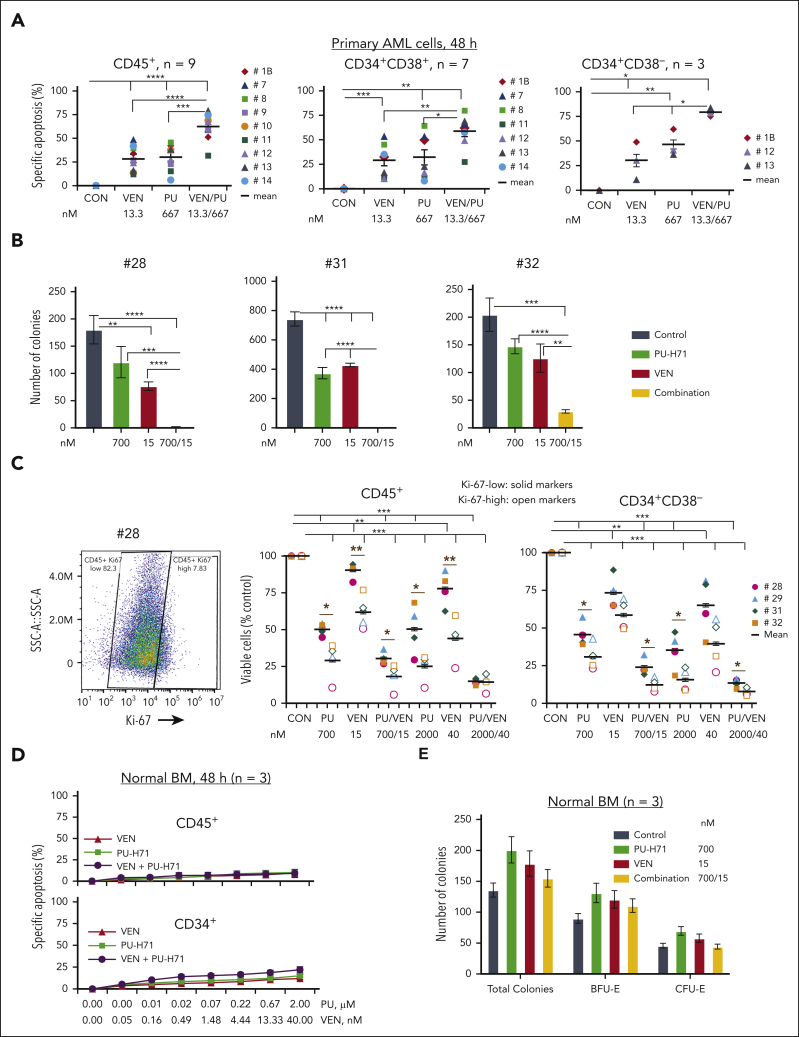
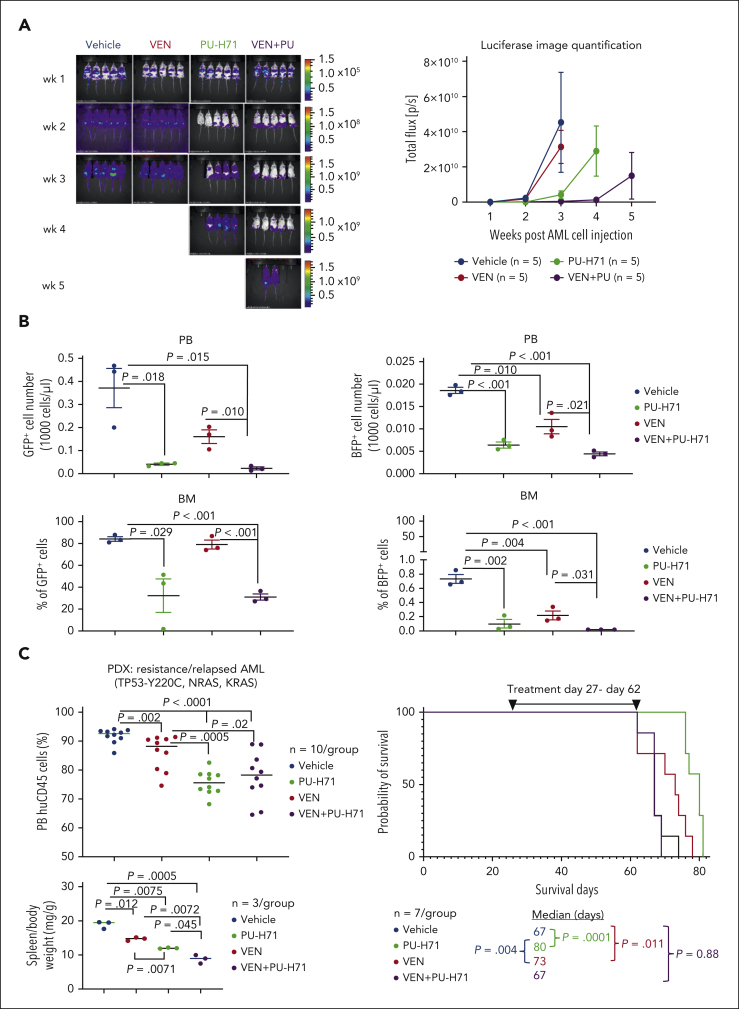
TP 53-mutant acute myeloid leukemia (AML) remains the ultimate therapeutic challenge. Epichaperomes, formed in malignant cells, consist of heat shock protein 90 (HSP90) and associated proteins that support the maturation, activity, and stability of oncogenic kinases and transcription factors including mutant p53. High-throughput drug screening identified HSP90 inhibitors as top hits in isogenic TP53-wild-type (WT) and -mutant AML cells. We detected epichaperomes in AML cells and stem/progenitor cells with TP53 mutations but not in healthy bone marrow (BM) cells. Hence, we investigated the therapeutic potential of specifically targeting epichaperomes with PU-H71 in TP53-mutant AML based on its preferred binding to HSP90 within epichaperomes. PU-H71 effectively suppressed cell intrinsic stress responses and killed AML cells, primarily by inducing apoptosis; targeted TP53-mutant stem/progenitor cells; and prolonged survival of TP53-mutant AML xenograft and patient-derived xenograft models, but it had minimal effects on healthy human BM CD34+ cells or on murine hematopoiesis. PU-H71 decreased MCL-1 and multiple signal proteins, increased proapoptotic Bcl-2-like protein 11 levels, and synergized with BCL-2 inhibitor venetoclax in TP53-mutant AML. Notably, PU-H71 effectively killed TP53-WT and -mutant cells in isogenic TP53-WT/TP53-R248W Molm13 cell mixtures, whereas MDM2 or BCL-2 inhibition only reduced TP53-WT but favored the outgrowth of TP53-mutant cells. Venetoclax enhanced the killing of both TP53-WT and -mutant cells by PU-H71 in a xenograft model. Our data suggest that epichaperome function is essential for TP53-mutant AML growth and survival and that its inhibition targets mutant AML and stem/progenitor cells, enhances venetoclax activity, and prevents the outgrowth of venetoclax-resistant TP53-mutant AML clones. These concepts warrant clinical evaluation.
© 2023 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: B.W. and R.A.M. are employees of Samus Therapeutics Inc. B.L.E. has received research funding from Celgene, Deerfield, Novartis, and Calico and consulting fees from GRAIL; he is a member of the scientific advisory board and shareholder for Neomorph Inc, TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Targeting the epichaperome to combat AML.Blood. 2023 Sep 21;142(12):1031-1032. doi: 10.1182/blood.2023021386. Blood. 2023. PMID: 37733379 No abstract available.
References
-
- Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
