

Mechanisms of readthrough mitigation reveal principles of GCN1-mediated translational quality control
- PMID: 37339632
- PMCID: PMC10364623
- DOI: 10.1016/j.cell.2023.05.035
Mechanisms of readthrough mitigation reveal principles of GCN1-mediated translational quality control
Abstract
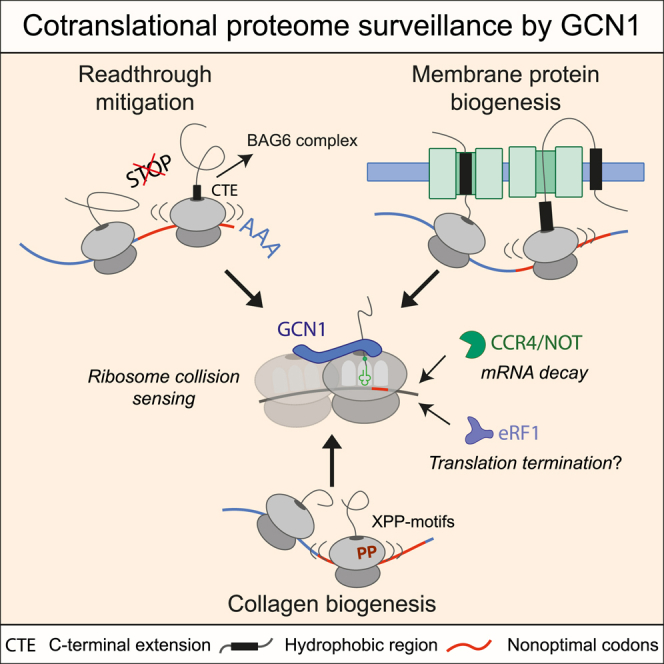
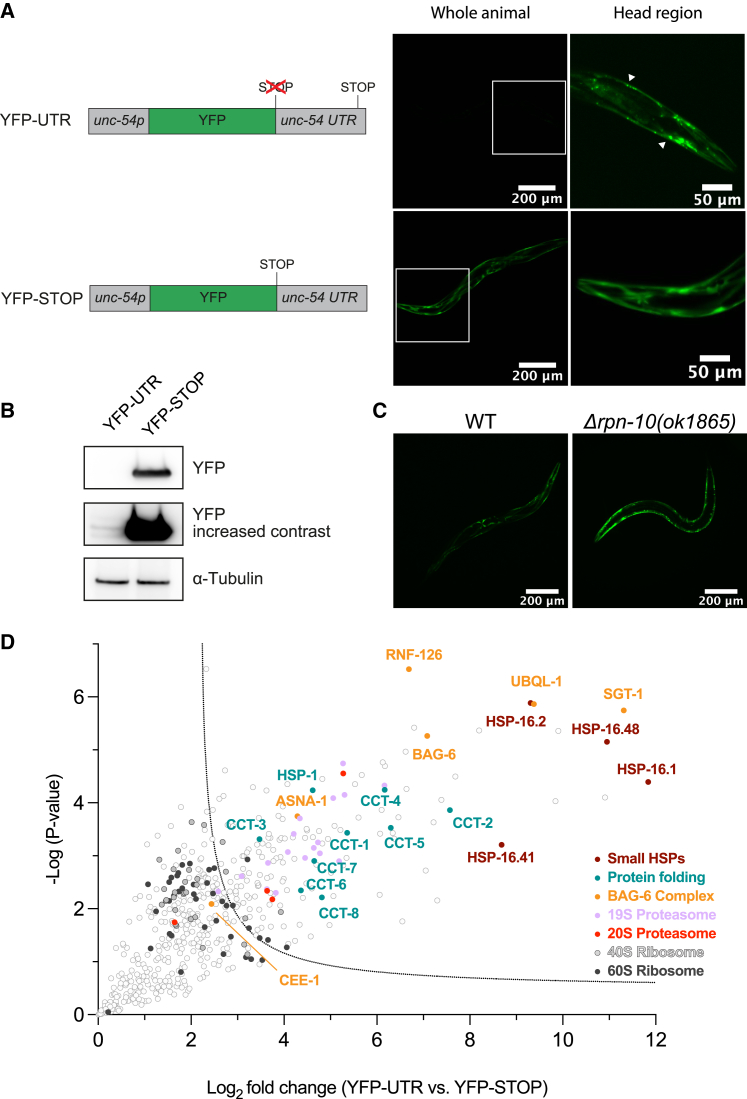
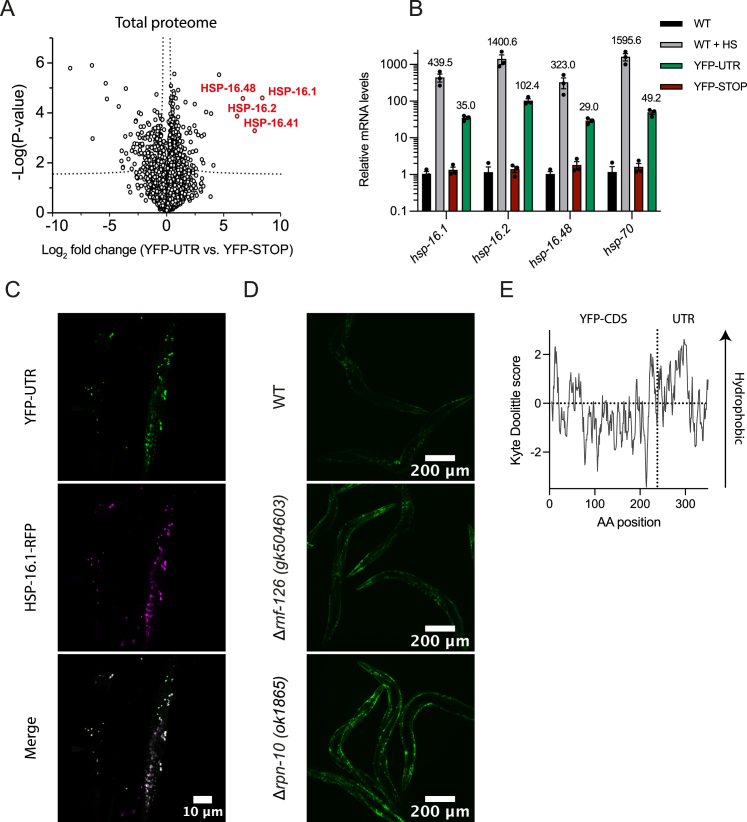
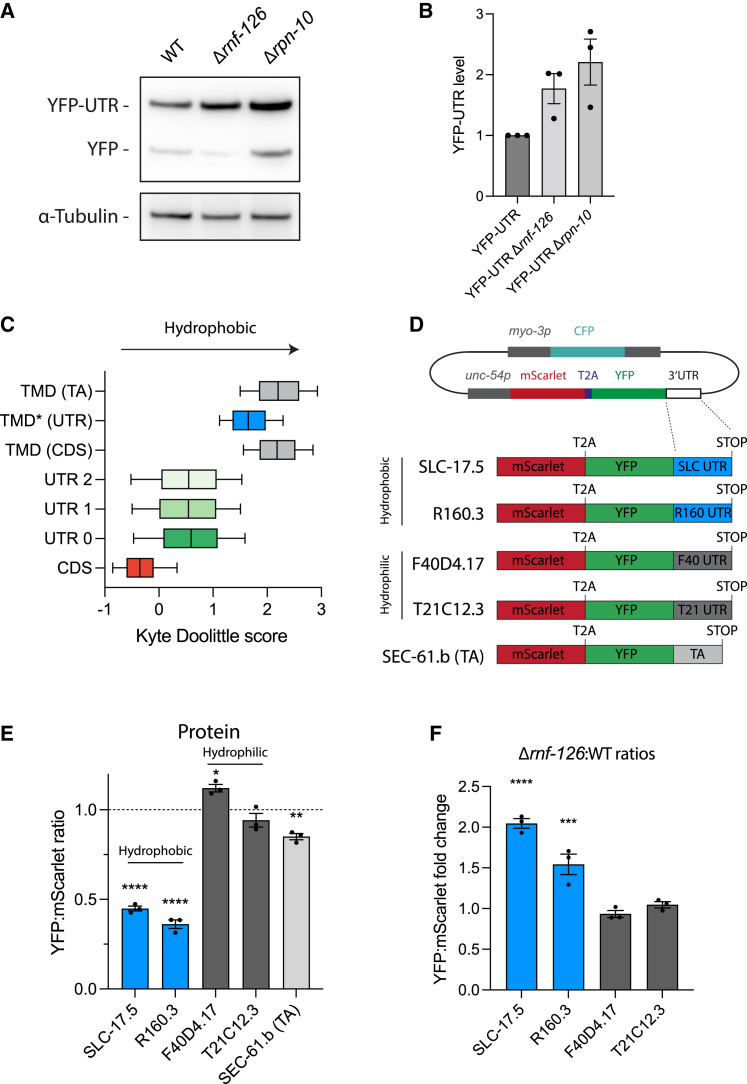
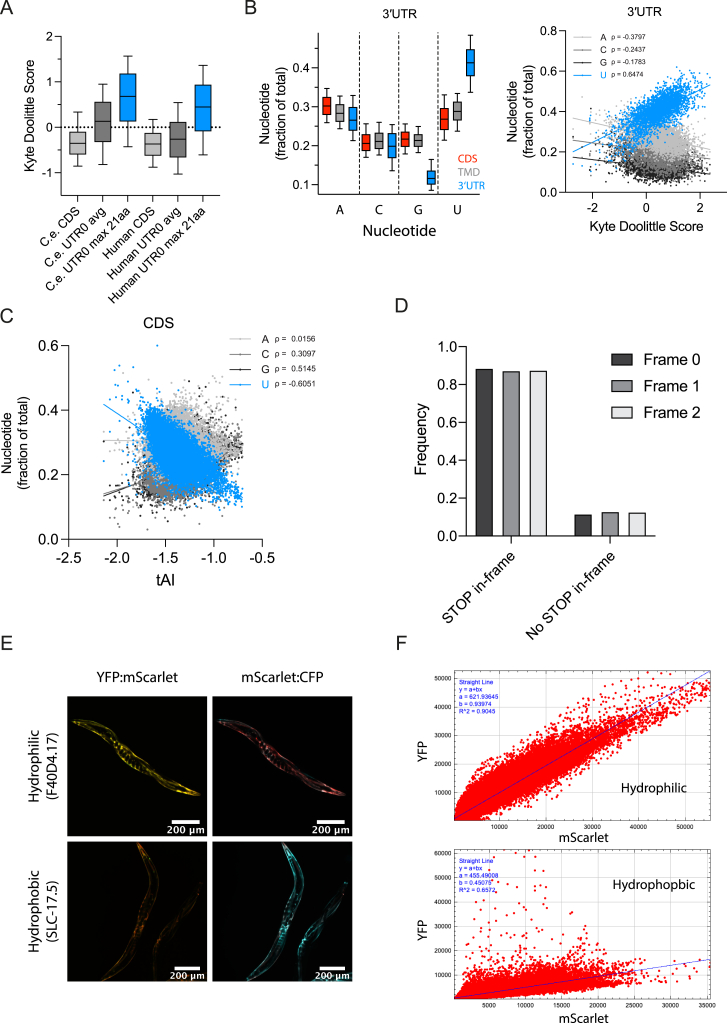
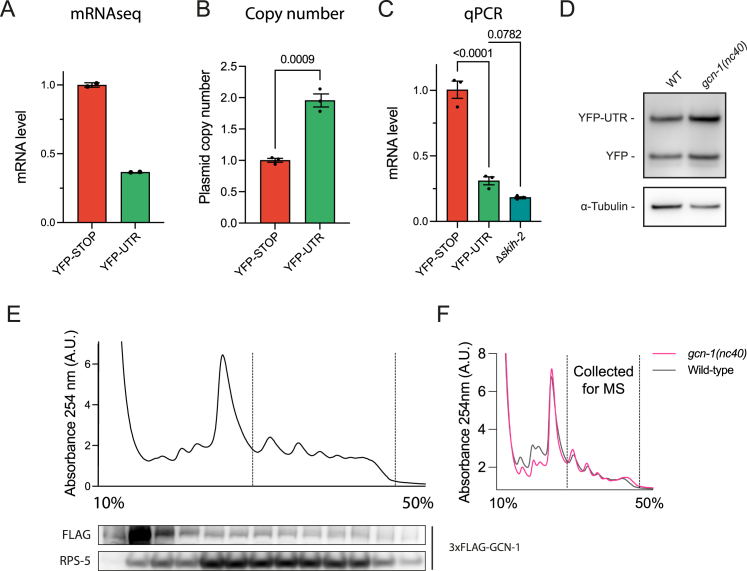
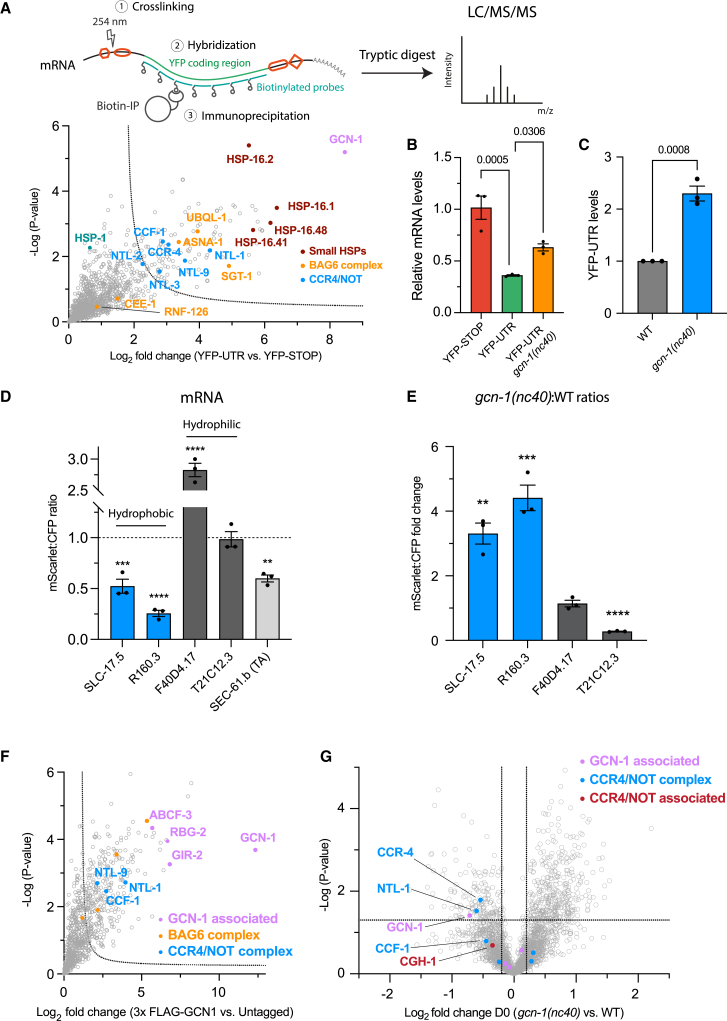
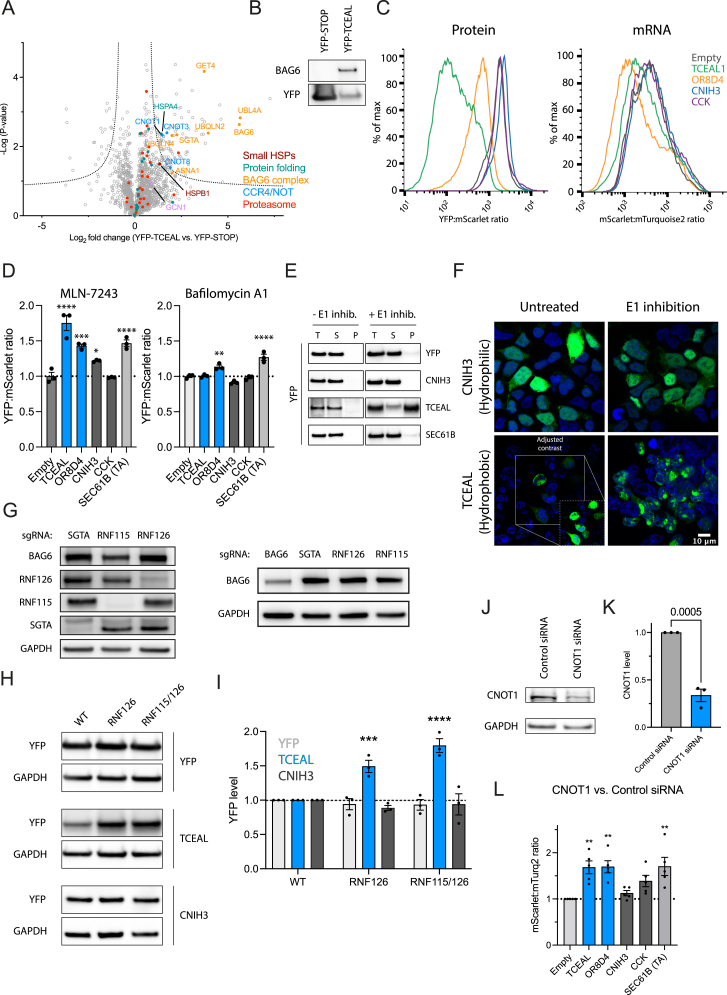
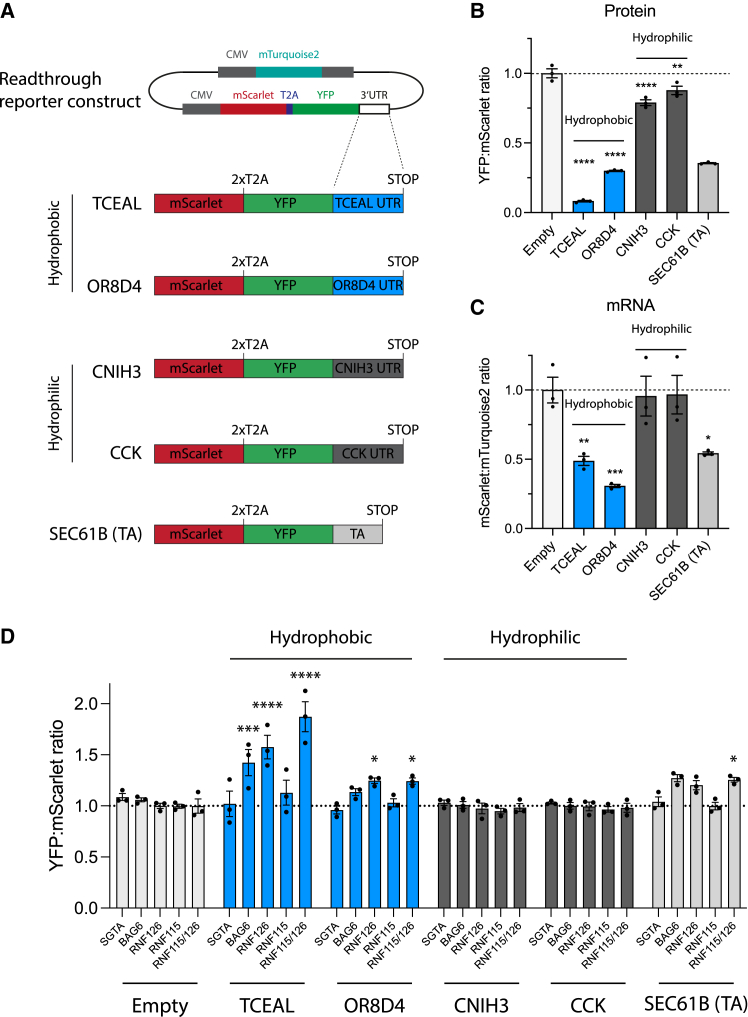
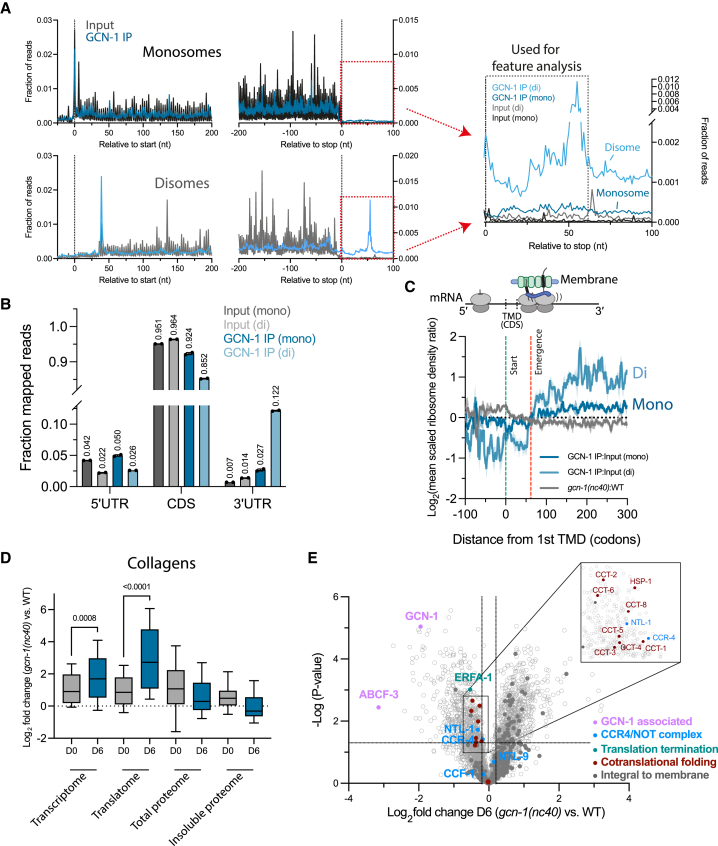
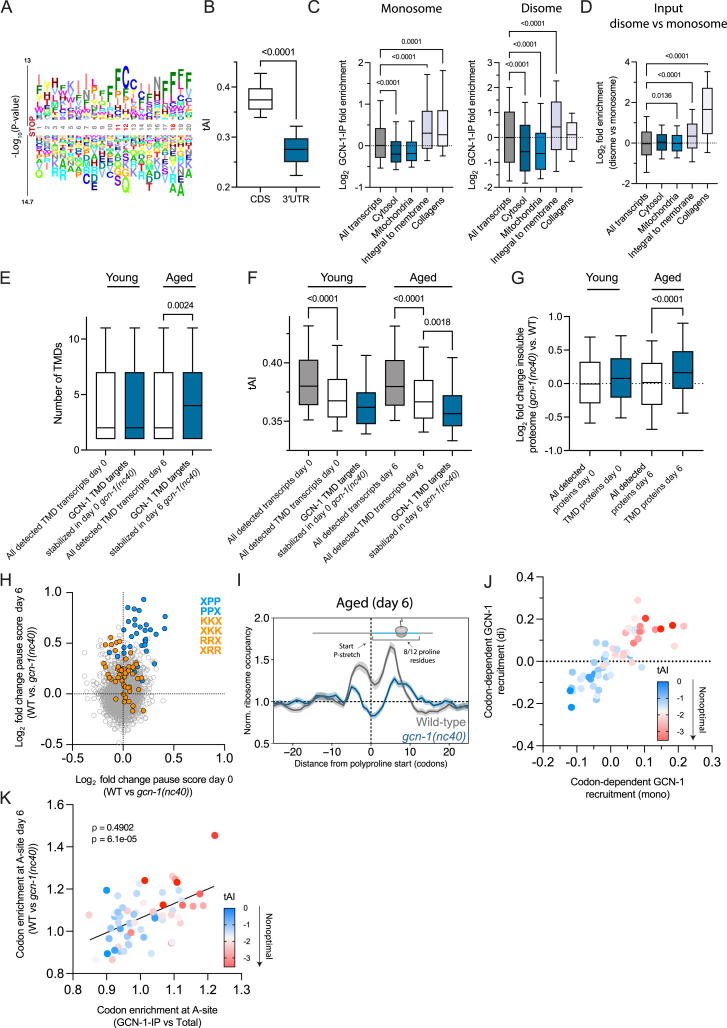
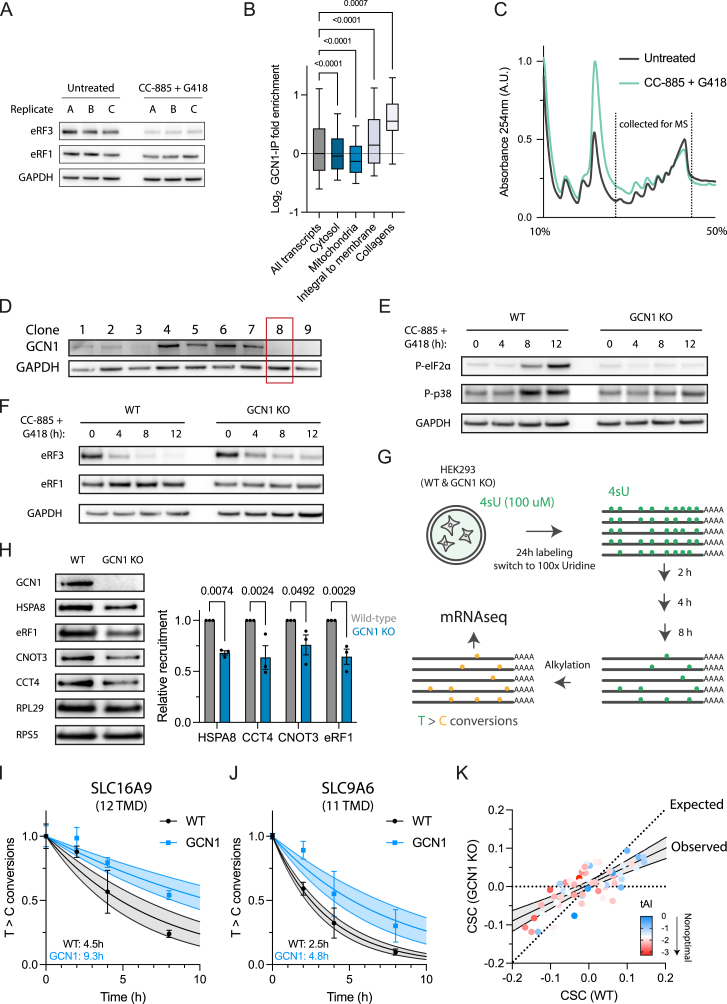
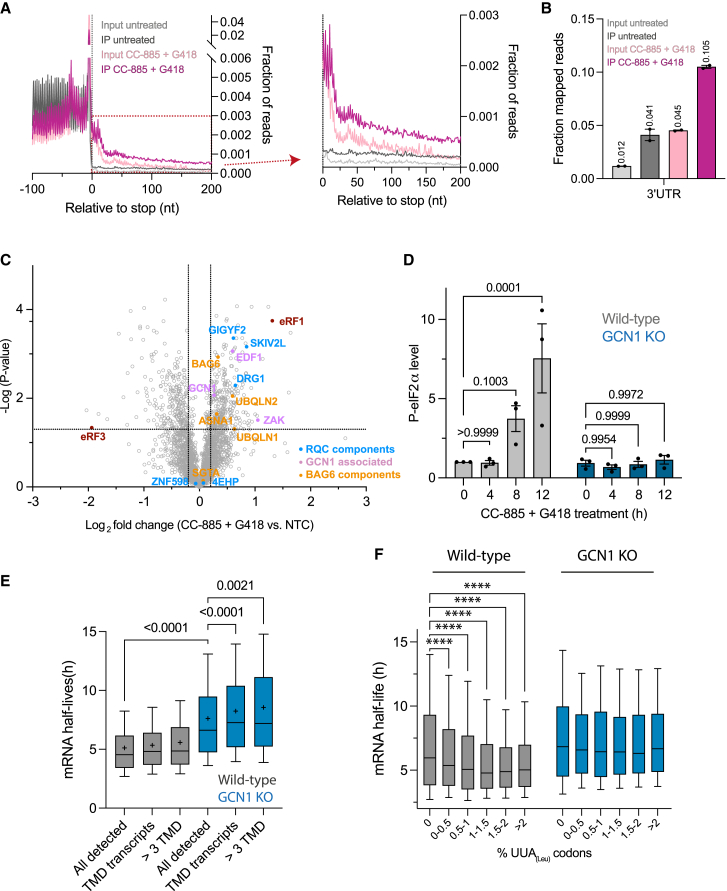
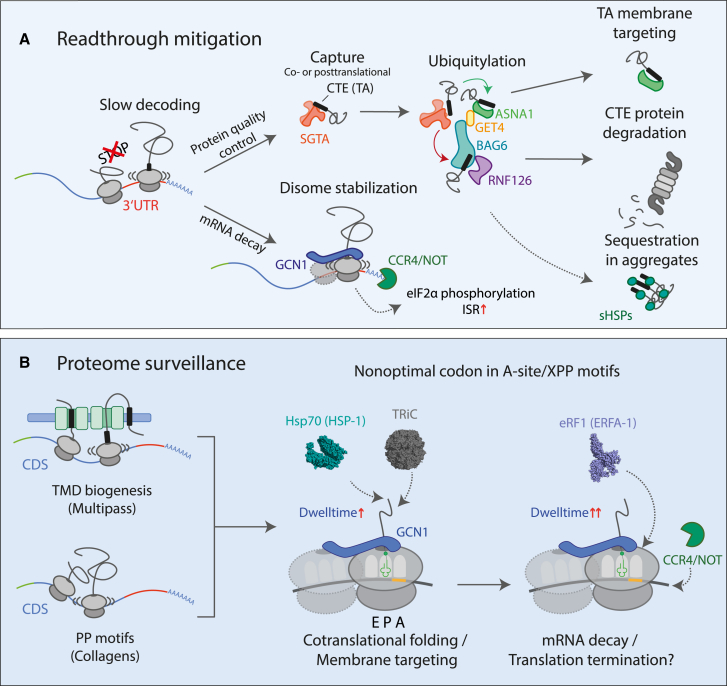
Readthrough into the 3' untranslated region (3' UTR) of the mRNA results in the production of aberrant proteins. Metazoans efficiently clear readthrough proteins, but the underlying mechanisms remain unknown. Here, we show in Caenorhabditis elegans and mammalian cells that readthrough proteins are targeted by a coupled, two-level quality control pathway involving the BAG6 chaperone complex and the ribosome-collision-sensing protein GCN1. Readthrough proteins with hydrophobic C-terminal extensions (CTEs) are recognized by SGTA-BAG6 and ubiquitylated by RNF126 for proteasomal degradation. Additionally, cotranslational mRNA decay initiated by GCN1 and CCR4/NOT limits the accumulation of readthrough products. Unexpectedly, selective ribosome profiling uncovered a general role of GCN1 in regulating translation dynamics when ribosomes collide at nonoptimal codons, enriched in 3' UTRs, transmembrane proteins, and collagens. GCN1 dysfunction increasingly perturbs these protein classes during aging, resulting in mRNA and proteome imbalance. Our results define GCN1 as a key factor acting during translation in maintaining protein homeostasis.
Keywords: BAG6 complex; CCR4/NOT; GCN1; aging; codon optimality; collagens; cotranslational mRNA decay; disomes; readthrough mitigation; transmembrane proteins.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
