

Epistatic interactions between the high pathogenicity island and other iron uptake systems shape Escherichia coli extra-intestinal virulence
- PMID: 37339949
- PMCID: PMC10282060
- DOI: 10.1038/s41467-023-39428-y
Epistatic interactions between the high pathogenicity island and other iron uptake systems shape Escherichia coli extra-intestinal virulence
Abstract
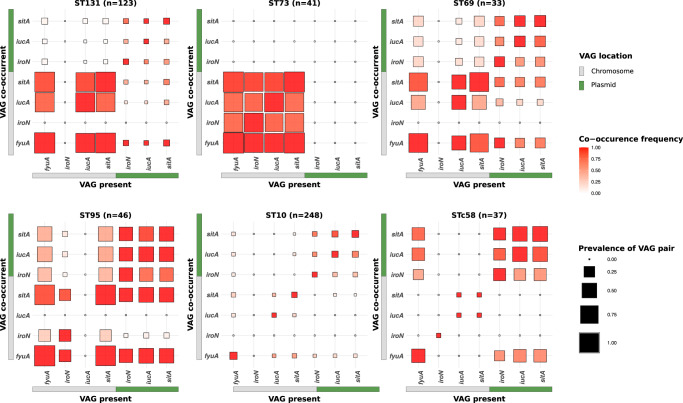
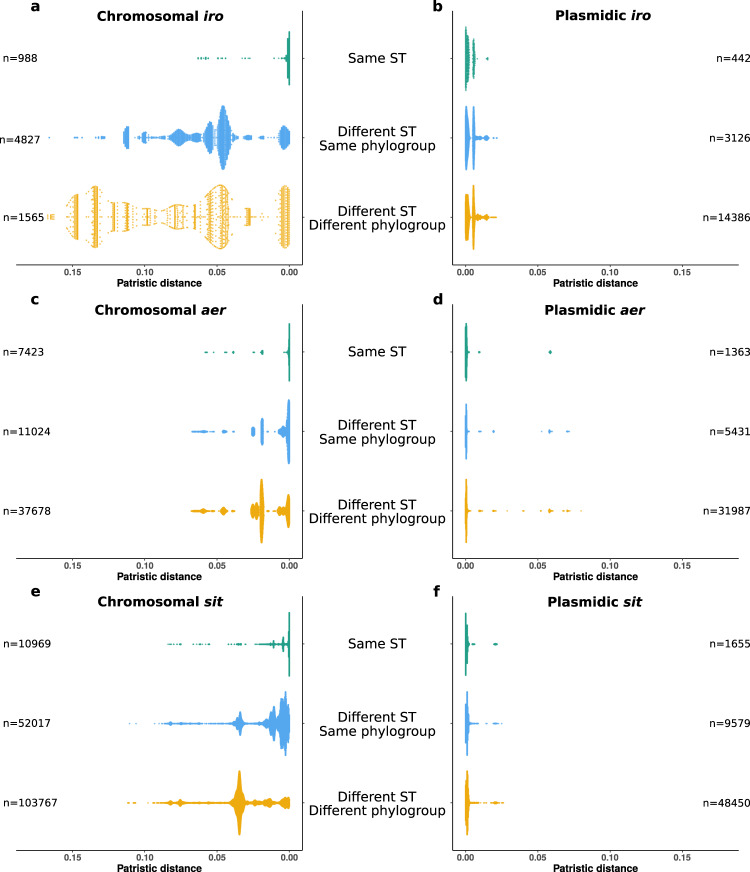
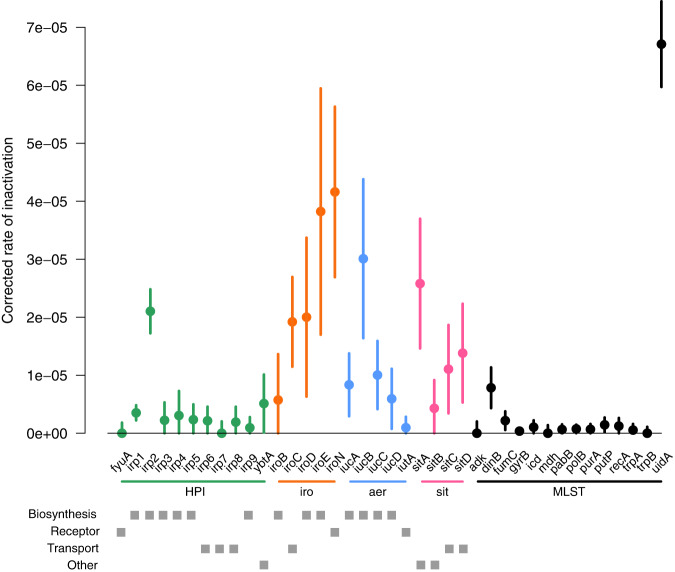
The intrinsic virulence of extra-intestinal pathogenic Escherichia coli is associated with numerous chromosomal and/or plasmid-borne genes, encoding diverse functions such as adhesins, toxins, and iron capture systems. However, the respective contribution to virulence of those genes seems to depend on the genetic background and is poorly understood. Here, we analyze genomes of 232 strains of sequence type complex STc58 and show that virulence (quantified in a mouse model of sepsis) emerged in a sub-group of STc58 due to the presence of the siderophore-encoding high-pathogenicity island (HPI). When extending our genome-wide association study to 370 Escherichia strains, we show that full virulence is associated with the presence of the aer or sit operons, in addition to the HPI. The prevalence of these operons, their co-occurrence and their genomic location depend on strain phylogeny. Thus, selection of lineage-dependent specific associations of virulence-associated genes argues for strong epistatic interactions shaping the emergence of virulence in E. coli.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Major role of iron uptake systems in the intrinsic extra-intestinal virulence of the genus Escherichia revealed by a genome-wide association study.PLoS Genet. 2020 Oct 28;16(10):e1009065. doi: 10.1371/journal.pgen.1009065. eCollection 2020 Oct. PLoS Genet. 2020. PMID: 33112851 Free PMC article.
-
Strain-specific impact of the high-pathogenicity island on virulence in extra-intestinal pathogenic Escherichia coli.Int J Med Microbiol. 2017 Jan;307(1):44-56. doi: 10.1016/j.ijmm.2016.11.004. Epub 2016 Nov 22. Int J Med Microbiol. 2017. PMID: 27923724
-
Evolutionary Dynamics Based on Comparative Genomics of Pathogenic Escherichia coli Lineages Harboring Polyketide Synthase (pks) Island.mBio. 2021 Mar 2;12(1):e03634-20. doi: 10.1128/mBio.03634-20. mBio. 2021. PMID: 33653937 Free PMC article.
-
The population genetics of pathogenic Escherichia coli.Nat Rev Microbiol. 2021 Jan;19(1):37-54. doi: 10.1038/s41579-020-0416-x. Epub 2020 Aug 21. Nat Rev Microbiol. 2021. PMID: 32826992 Review.
-
Enterohaemorrhagic Escherichia coli O26:H11/H-: a human pathogen in emergence.Berl Munch Tierarztl Wochenschr. 2007 Jul-Aug;120(7-8):279-87. Berl Munch Tierarztl Wochenschr. 2007. PMID: 17715820 Review.
Cited by
-
Developing the PIP-eco: An integrated genomic pipeline for identification and characterization of Escherichia coli pathotypes encompassing hybrid forms.Comput Struct Biotechnol J. 2024 Jul 20;23:3040-3049. doi: 10.1016/j.csbj.2024.07.017. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 39175796 Free PMC article.
-
National genomic epidemiology investigation revealed the spread of carbapenem-resistant Escherichia coli in healthy populations and the impact on public health.Genome Med. 2024 Apr 16;16(1):57. doi: 10.1186/s13073-024-01310-x. Genome Med. 2024. PMID: 38627827 Free PMC article.
-
Bloodstream infections: mechanisms of pathogenesis and opportunities for intervention.Nat Rev Microbiol. 2025 Apr;23(4):210-224. doi: 10.1038/s41579-024-01105-2. Epub 2024 Oct 17. Nat Rev Microbiol. 2025. PMID: 39420097 Review.
-
A genome-wide association study reveals that epistasis underlies the pathogenicity of Pectobacterium.Microbiol Spectr. 2023 Sep 15;11(5):e0176423. doi: 10.1128/spectrum.01764-23. Online ahead of print. Microbiol Spectr. 2023. PMID: 37712699 Free PMC article.
-
Major F plasmid clusters are linked with ColV and pUTI89-like marker genes in bloodstream isolates of Escherichia coli.BMC Genomics. 2025 Jan 21;26(1):57. doi: 10.1186/s12864-025-11226-4. BMC Genomics. 2025. PMID: 39838323 Free PMC article.
References
-
- Desjardins P, Picard B, Kaltenböck B, Elion J, Denamur E. Sex in Escherichia coli does not disrupt the clonal structure of the population: evidence from random amplified polymorphic DNA and restriction-fragment-length polymorphism. J. Mol. Evol. 1995;41:440–448. doi: 10.1007/BF00160315. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
