

Imputation of ancient human genomes
- PMID: 37339987
- PMCID: PMC10282092
- DOI: 10.1038/s41467-023-39202-0
Imputation of ancient human genomes
Abstract
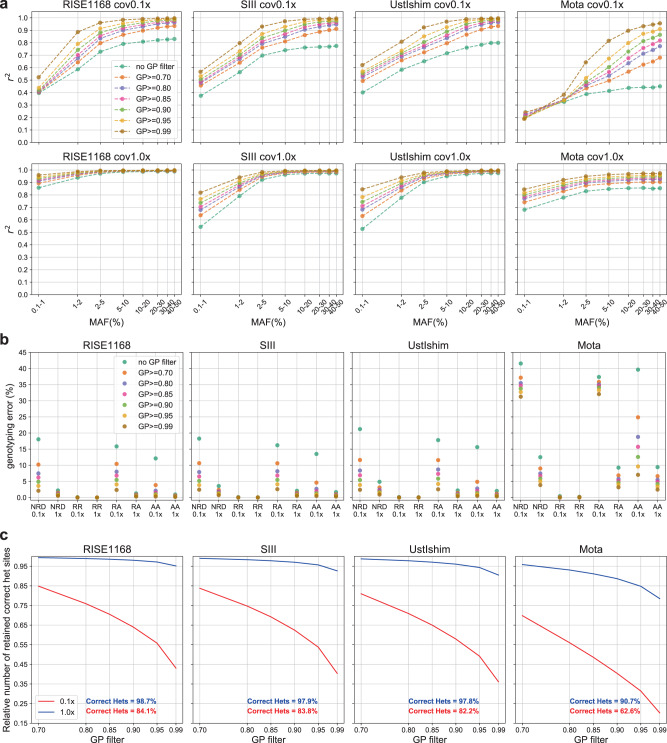
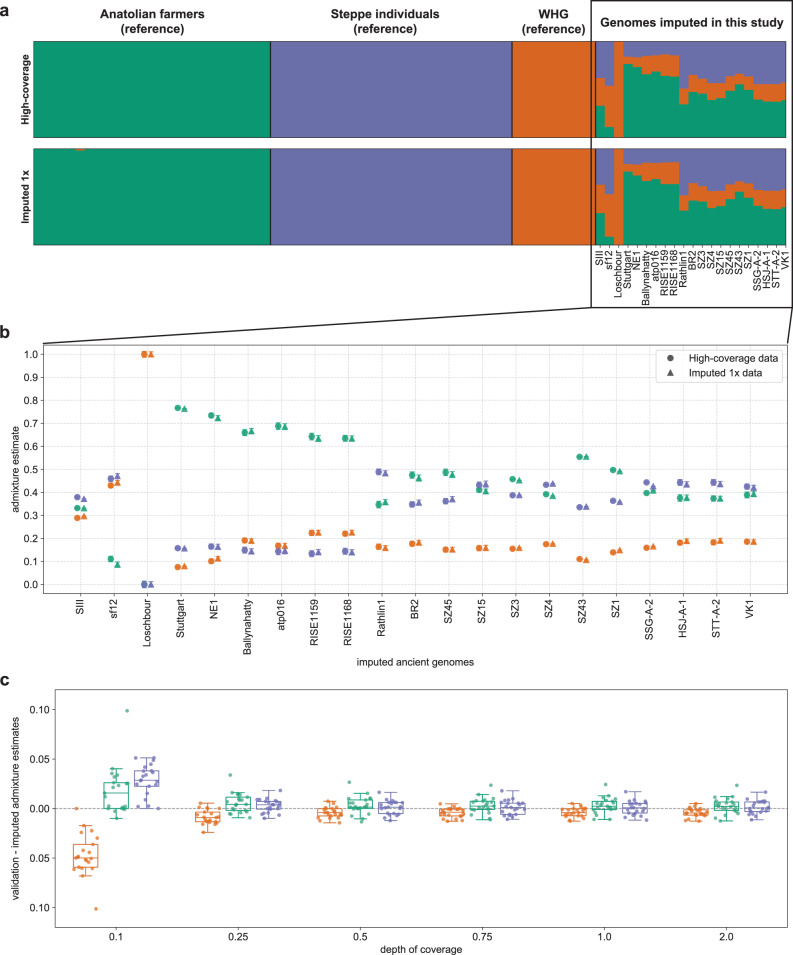
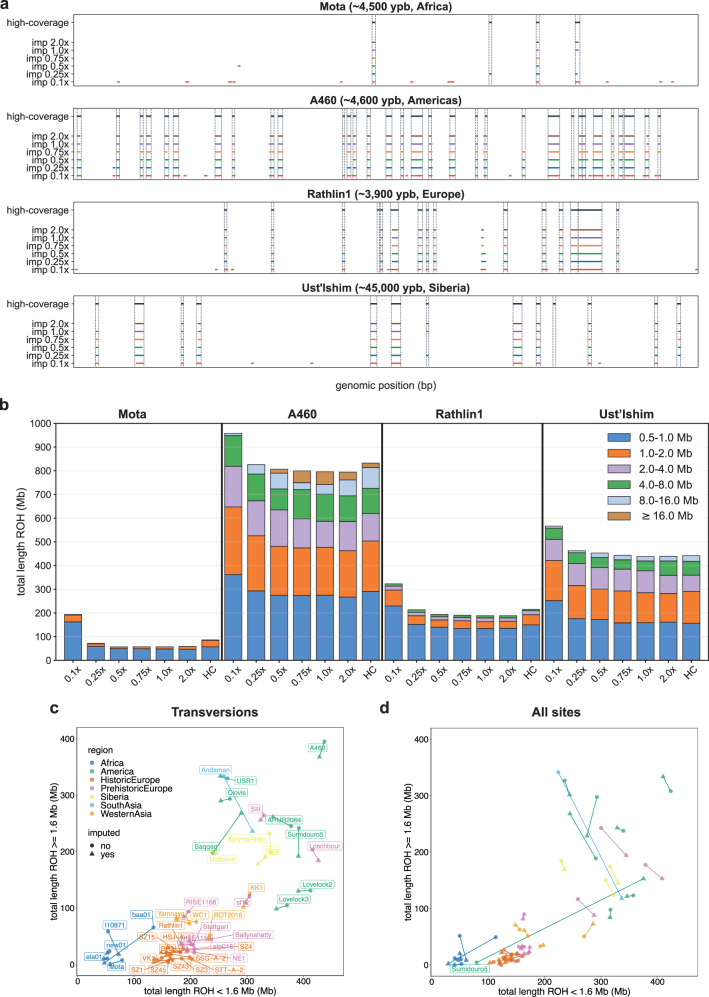
Due to postmortem DNA degradation and microbial colonization, most ancient genomes have low depth of coverage, hindering genotype calling. Genotype imputation can improve genotyping accuracy for low-coverage genomes. However, it is unknown how accurate ancient DNA imputation is and whether imputation introduces bias to downstream analyses. Here we re-sequence an ancient trio (mother, father, son) and downsample and impute a total of 43 ancient genomes, including 42 high-coverage (above 10x) genomes. We assess imputation accuracy across ancestries, time, depth of coverage, and sequencing technology. We find that ancient and modern DNA imputation accuracies are comparable. When downsampled at 1x, 36 of the 42 genomes are imputed with low error rates (below 5%) while African genomes have higher error rates. We validate imputation and phasing results using the ancient trio data and an orthogonal approach based on Mendel's rules of inheritance. We further compare the downstream analysis results between imputed and high-coverage genomes, notably principal component analysis, genetic clustering, and runs of homozygosity, observing similar results starting from 0.5x coverage, except for the African genomes. These results suggest that, for most populations and depths of coverage as low as 0.5x, imputation is a reliable method that can improve ancient DNA studies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Günther, T. & Jakobsson, M. Population genomic analyses of DNA from ancient remains. In: Handbook of Statistical Genomics1, 295–324 (Wiley, 2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
