

Differential dysregulation of β-TrCP1 and -2 by HIV-1 Vpu leads to inhibition of canonical and non-canonical NF-κB pathways in infected cells
- PMID: 37341489
- PMCID: PMC10470808
- DOI: 10.1128/mbio.03293-22
Differential dysregulation of β-TrCP1 and -2 by HIV-1 Vpu leads to inhibition of canonical and non-canonical NF-κB pathways in infected cells
Abstract
The HIV-1 Vpu protein is expressed late in the virus lifecycle to promote infectious virus production and avoid innate and adaptive immunity. This includes the inhibition of the NF-κB pathway which, when activated, leads to the induction of inflammatory responses and the promotion of antiviral immunity. Here we demonstrate that Vpu can inhibit both canonical and non-canonical NF-κB pathways, through the direct inhibition of the F-box protein β-TrCP, the substrate recognition portion of the Skp1-Cul1-F-box (SCF)β-TrCP ubiquitin ligase complex. There are two paralogues of β-TrCP (β-TrCP1/BTRC and β-TrCP2/FBXW11), encoded on different chromosomes, which appear to be functionally redundant. Vpu, however, is one of the few β-TrCP substrates to differentiate between the two paralogues. We have found that patient-derived alleles of Vpu, unlike those from lab-adapted viruses, trigger the degradation of β-TrCP1 while co-opting its paralogue β-TrCP2 for the degradation of cellular targets of Vpu, such as CD4. The potency of this dual inhibition correlates with stabilization of the classical IκBα and the phosphorylated precursors of the mature DNA-binding subunits of canonical and non-canonical NF-κB pathways, p105/NFκB1 and p100/NFκB2, in HIV-1 infected CD4+ T cells. Both precursors act as alternative IκBs in their own right, thus reinforcing NF-κB inhibition at steady state and upon activation with either selective canonical or non-canonical NF-κB stimuli. These data reveal the complex regulation of NF-κB late in the viral replication cycle, with consequences for both the pathogenesis of HIV/AIDS and the use of NF-κB-modulating drugs in HIV cure strategies. IMPORTANCE The NF-κB pathway regulates host responses to infection and is a common target of viral antagonism. The HIV-1 Vpu protein inhibits NF-κB signaling late in the virus lifecycle, by binding and inhibiting β-TrCP, the substrate recognition portion of the ubiquitin ligase responsible for inducing IκB degradation. Here we demonstrate that Vpu simultaneously inhibits and exploits the two different paralogues of β-TrCP by triggering the degradation of β-TrCP1 and co-opting β-TrCP2 for the destruction of its cellular targets. In so doing, it has a potent inhibitory effect on both the canonical and non-canonical NF-κB pathways. This effect has been underestimated in previous mechanistic studies due to the use of Vpu proteins from lab-adapted viruses. Our findings reveal previously unappreciated differences in the β-TrCP paralogues, revealing functional insights into the regulation of these proteins. This study also raises important implications for the role of NF-κB inhibition in the immunopathogenesis of HIV/AIDS and the way that this may impact on HIV latency reversal strategies based on the activation of the non-canonical NF-κB pathway.
Keywords: HIV-1; Vpu; beta-TrCP; nuclear factor kappa B; ubiquitin ligase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
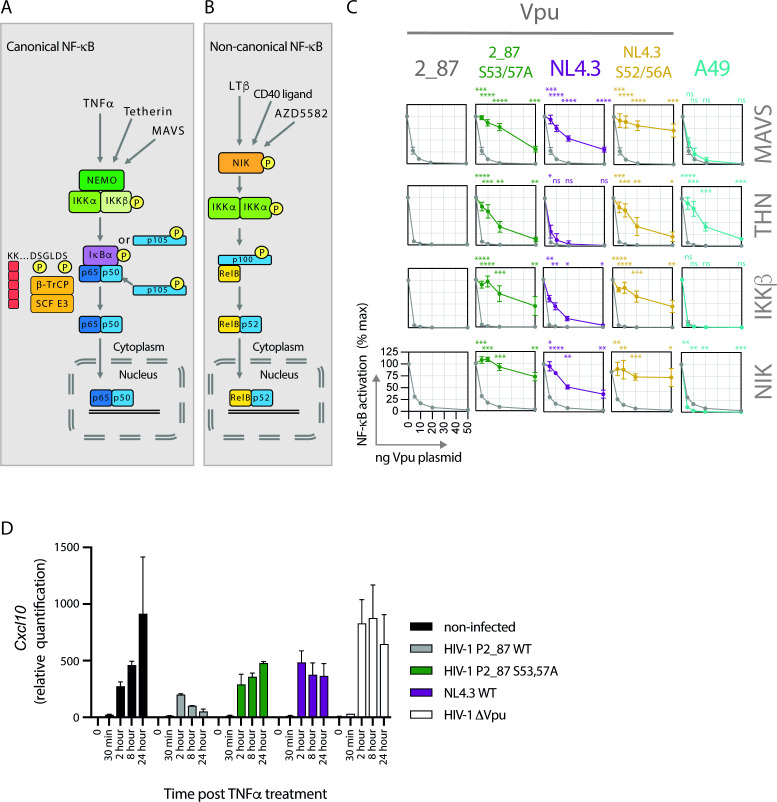
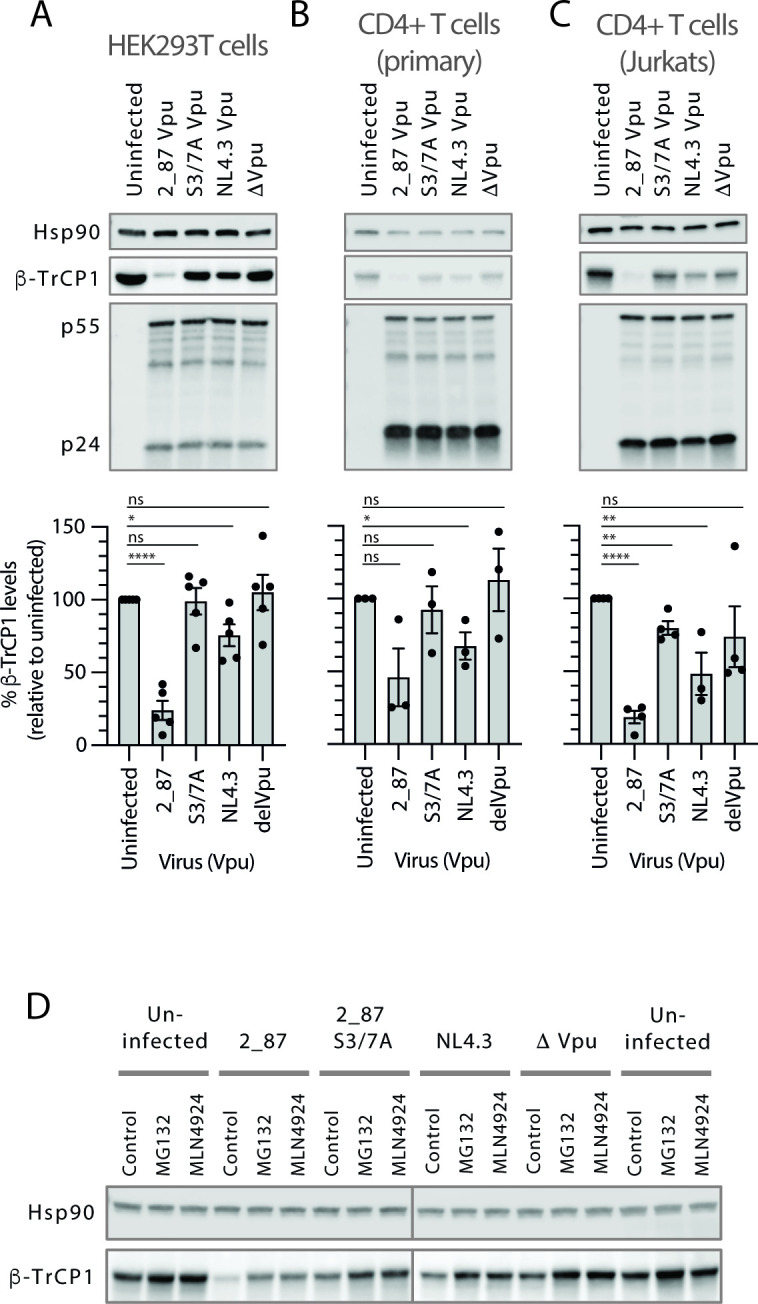
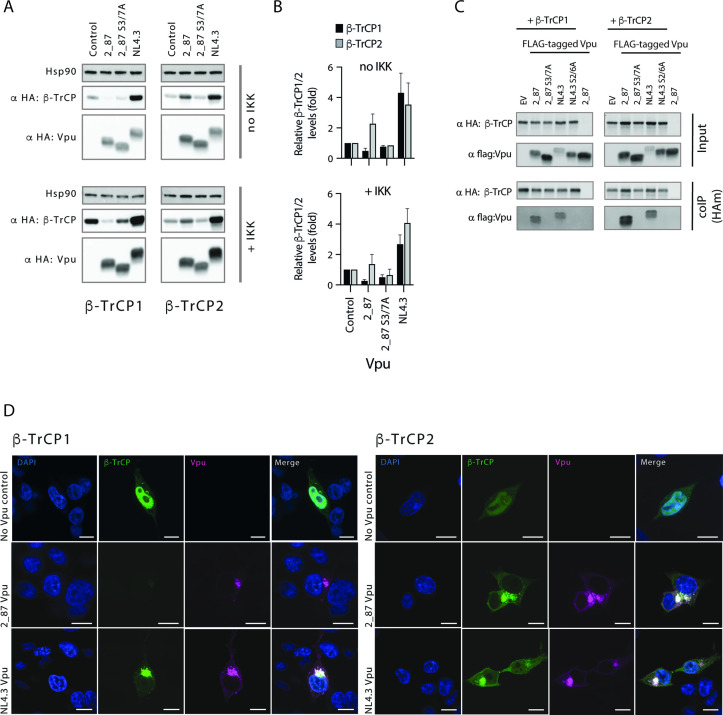
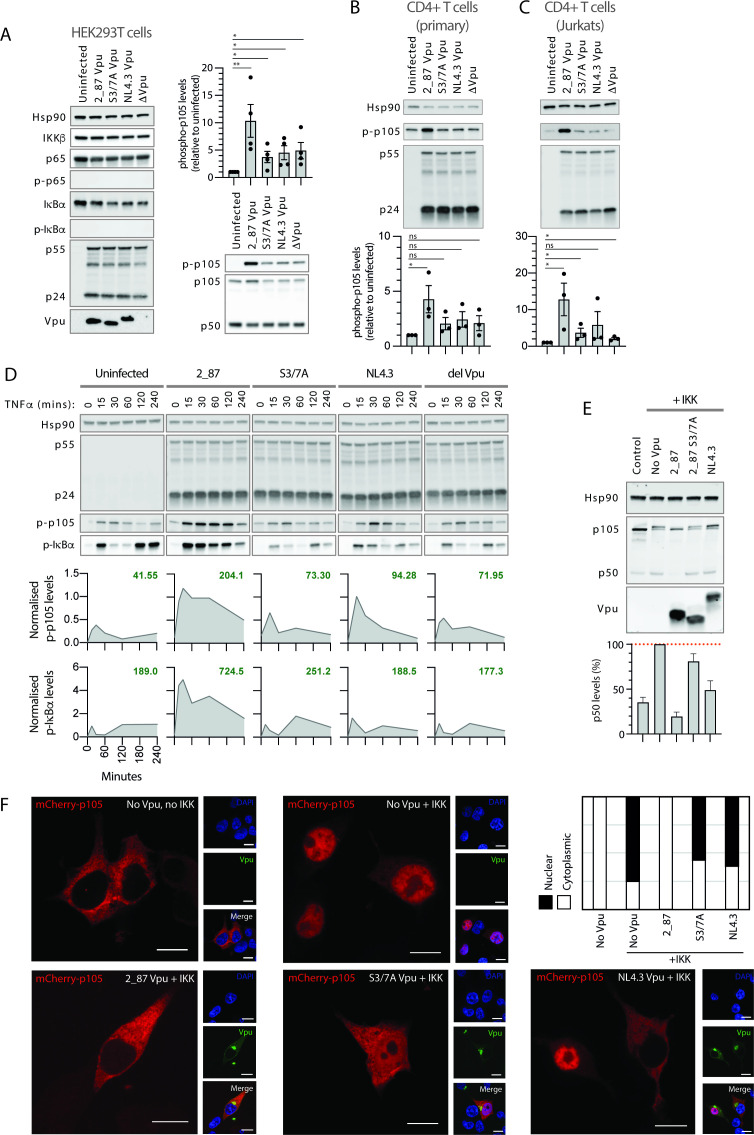
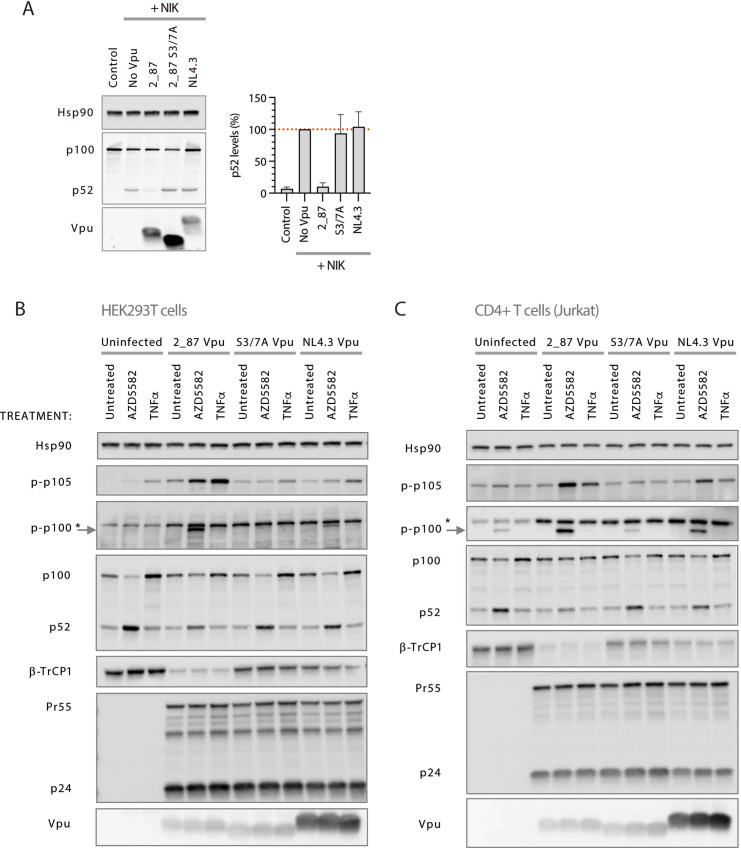
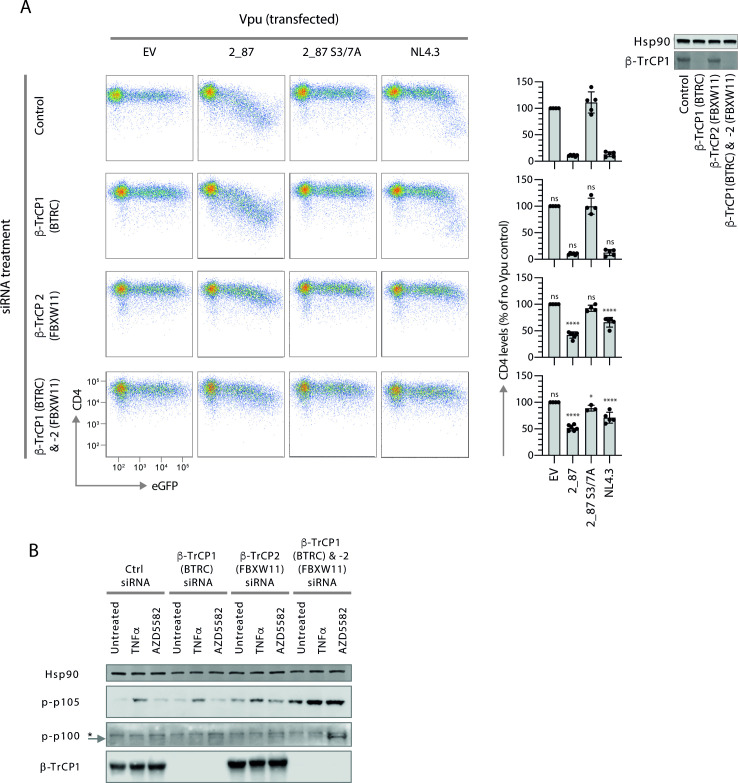
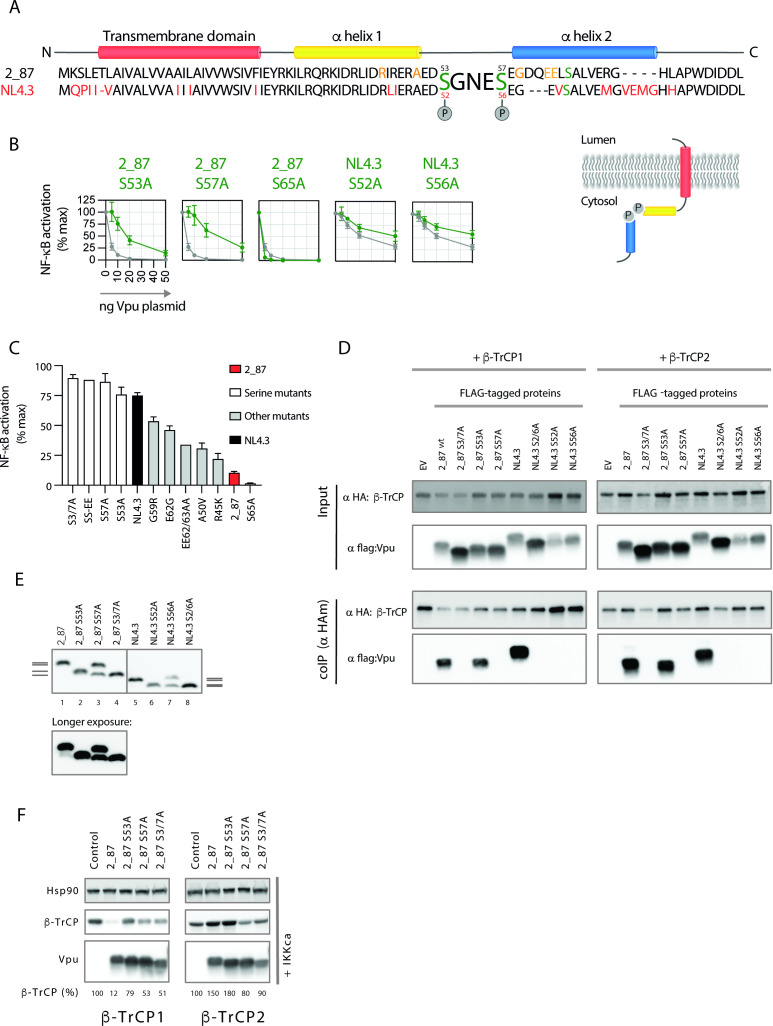
Comment in
-
Insights into the role of HIV-1 Vpu in modulation of NF-ĸB signaling pathways.mBio. 2023 Aug 31;14(4):e0092023. doi: 10.1128/mbio.00920-23. Epub 2023 Jul 6. mBio. 2023. PMID: 37409832 Free PMC article.
Similar articles
-
Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κB activation by using molecular mimicry to target β-TrCP.mBio. 2015 Jan 27;6(1):e02490-14. doi: 10.1128/mBio.02490-14. mBio. 2015. PMID: 25626907 Free PMC article.
-
The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B.J Biol Chem. 2001 May 11;276(19):15920-8. doi: 10.1074/jbc.M010533200. Epub 2001 Feb 16. J Biol Chem. 2001. PMID: 11278695
-
Inhibition of β-TrcP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells.Blood. 2011 Jun 16;117(24):6600-7. doi: 10.1182/blood-2011-01-333427. Epub 2011 Apr 26. Blood. 2011. PMID: 21521785
-
Regulation of NF-κB by ubiquitination and degradation of the IκBs.Immunol Rev. 2012 Mar;246(1):77-94. doi: 10.1111/j.1600-065X.2012.01098.x. Immunol Rev. 2012. PMID: 22435548 Review.
-
The characteristics and roles of β-TrCP1/2 in carcinogenesis.FEBS J. 2021 Jun;288(11):3351-3374. doi: 10.1111/febs.15585. Epub 2020 Oct 23. FEBS J. 2021. PMID: 33021036 Review.
Cited by
-
ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor.Biology (Basel). 2024 Feb 26;13(3):146. doi: 10.3390/biology13030146. Biology (Basel). 2024. PMID: 38534416 Free PMC article. Review.
-
SKP1-CUL1-F-box: Key molecular targets affecting disease progression.FASEB J. 2025 Jan 31;39(2):e70326. doi: 10.1096/fj.202402816RR. FASEB J. 2025. PMID: 39812503 Free PMC article. Review.
-
The African swine fever virus MGF300-4L protein is associated with viral pathogenicity by promoting the autophagic degradation of IKKβ and increasing the stability of IκBα.Emerg Microbes Infect. 2024 Dec;13(1):2333381. doi: 10.1080/22221751.2024.2333381. Epub 2024 Apr 14. Emerg Microbes Infect. 2024. PMID: 38501350 Free PMC article.
-
Insights into the role of HIV-1 Vpu in modulation of NF-ĸB signaling pathways.mBio. 2023 Aug 31;14(4):e0092023. doi: 10.1128/mbio.00920-23. Epub 2023 Jul 6. mBio. 2023. PMID: 37409832 Free PMC article.
-
HIV-1 Vpu interacts with RBM10 to promote HIV-1 infection.mSystems. 2025 Aug 19;10(8):e0040325. doi: 10.1128/msystems.00403-25. Epub 2025 Jul 31. mSystems. 2025. PMID: 40742131 Free PMC article.
References
-
- Khan H, Sumner RP, Rasaiyaah J, Tan CP, Rodriguez-Plata MT, Van Tulleken C, Fink D, Zuliani-Alvarez L, Thorne L, Stirling D, Milne RS, Towers GJ. 2020. HIV-1 Vpr Antagonizes innate immune activation by targeting karyopherin-mediated NF-ΚB/Irf3 nuclear transport. Elife 9:e60821. doi: 10.7554/eLife.60821 - DOI - PMC - PubMed
-
- Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, Walker BD, Thomas R, Shaw GM, Hahn BH, Keele BF, Lifson JD, Carrington M. 2016. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe 19:686–695. doi: 10.1016/j.chom.2016.04.005 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
