

Accelerating drug target inhibitor discovery with a deep generative foundation model
- PMID: 37343087
- PMCID: PMC10284550
- DOI: 10.1126/sciadv.adg7865
Accelerating drug target inhibitor discovery with a deep generative foundation model
Abstract
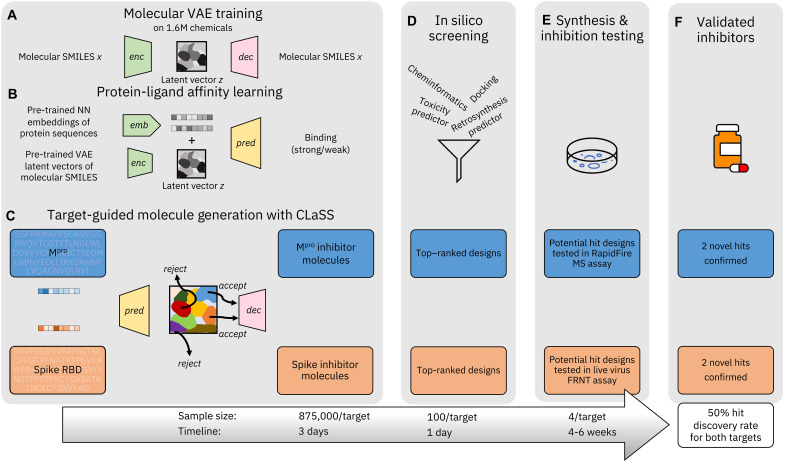
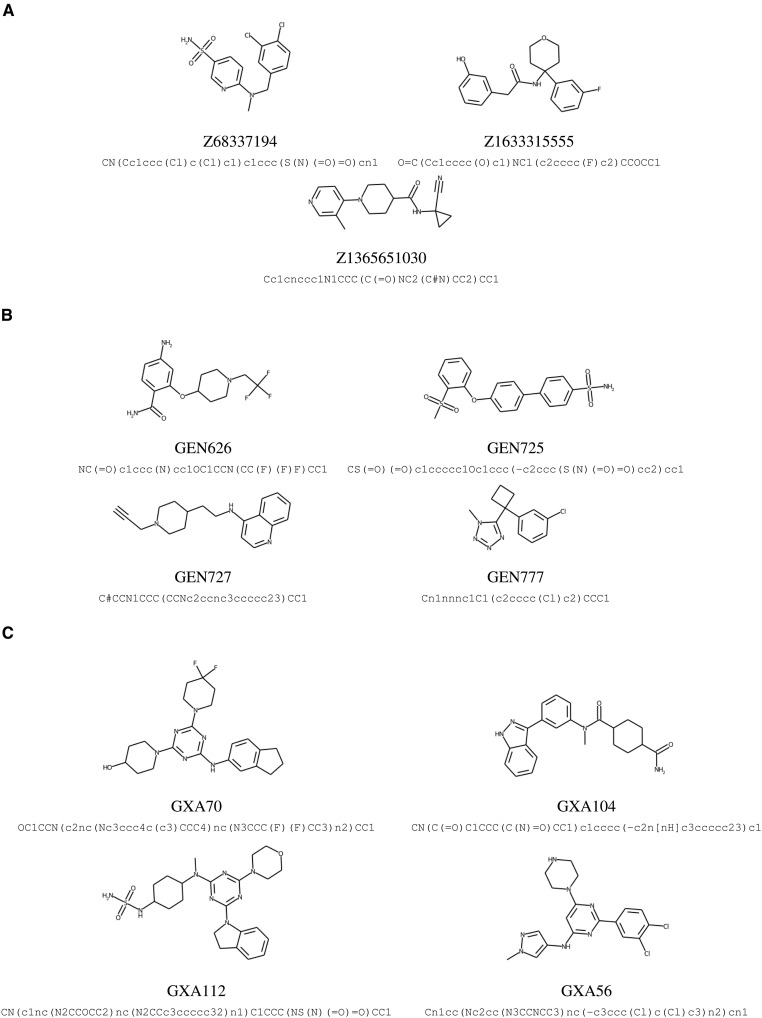
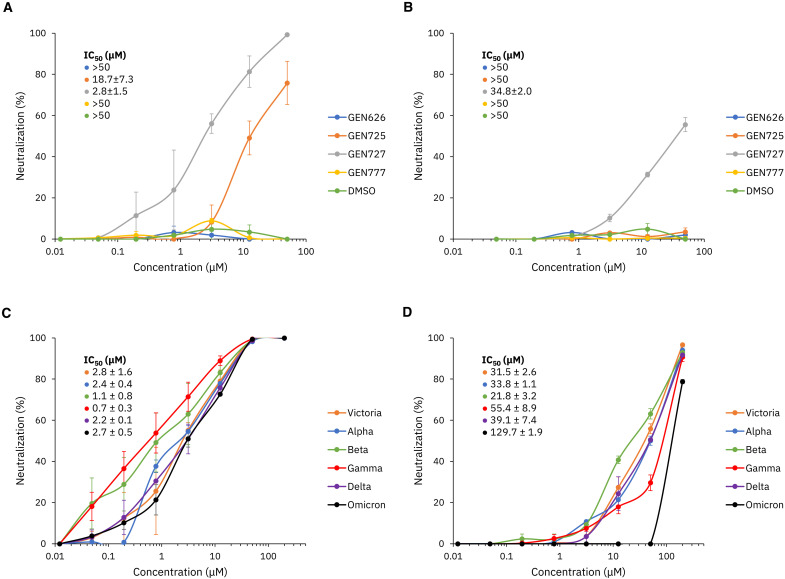
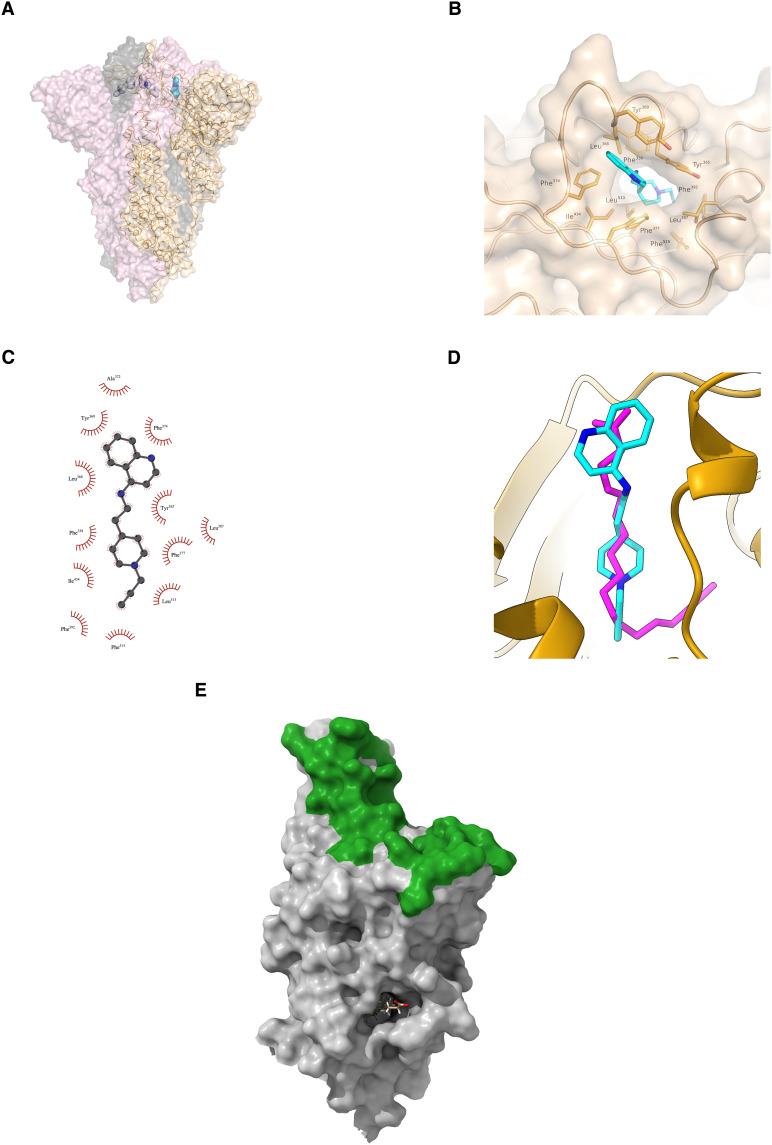
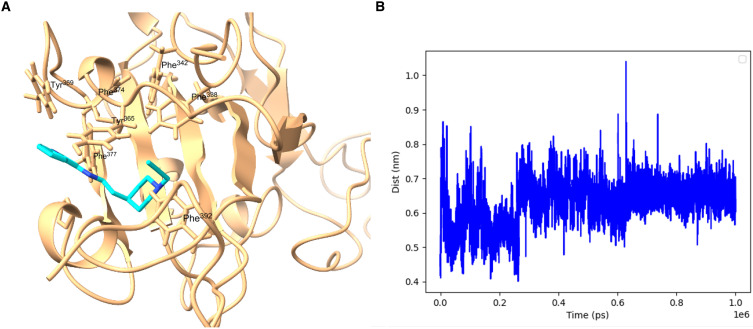
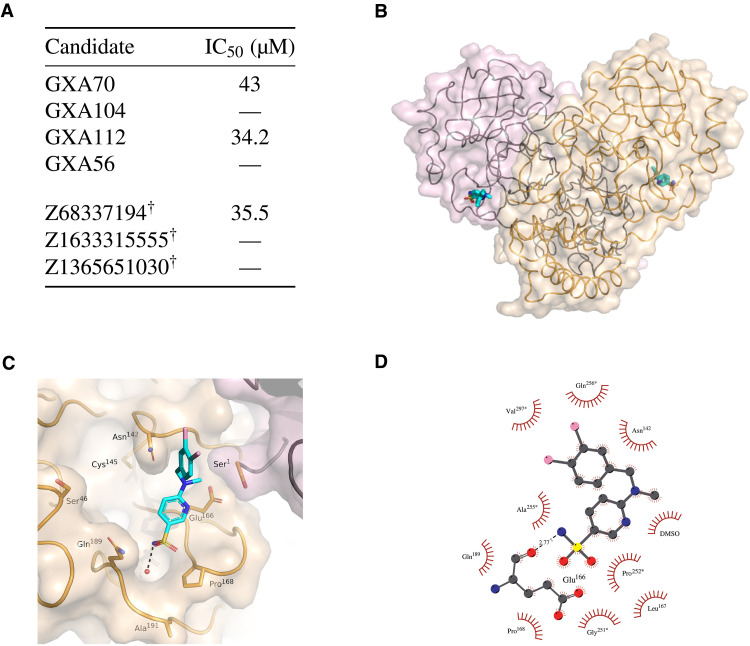
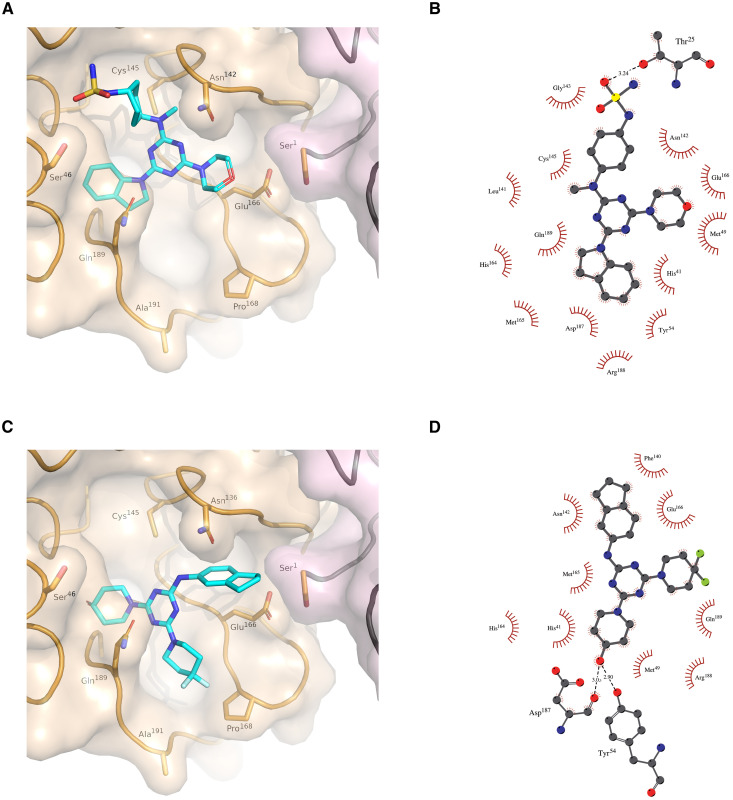
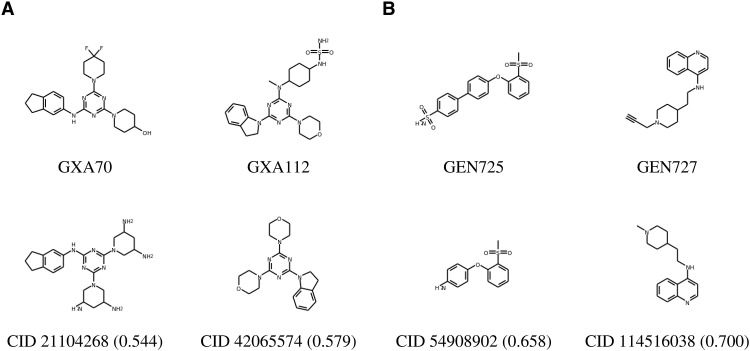
Inhibitor discovery for emerging drug-target proteins is challenging, especially when target structure or active molecules are unknown. Here, we experimentally validate the broad utility of a deep generative framework trained at-scale on protein sequences, small molecules, and their mutual interactions-unbiased toward any specific target. We performed a protein sequence-conditioned sampling on the generative foundation model to design small-molecule inhibitors for two dissimilar targets: the spike protein receptor-binding domain (RBD) and the main protease from SARS-CoV-2. Despite using only the target sequence information during the model inference, micromolar-level inhibition was observed in vitro for two candidates out of four synthesized for each target. The most potent spike RBD inhibitor exhibited activity against several variants in live virus neutralization assays. These results establish that a single, broadly deployable generative foundation model for accelerated inhibitor discovery is effective and efficient, even in the absence of target structure or binder information.
Figures
References
-
- M. D. Lloyd, High-throughput screening for the discovery of enzyme inhibitors. J. Med. Chem. 63, 10742–10772 (2020). - PubMed
-
- P. G. Polishchuk, T. I. Madzhidov, A. Varnek, Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 27, 675–679 (2013). - PubMed
-
- J. A. DiMasi, H. G. Grabowski, R. W. Hansen, Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20–33 (2016). - PubMed
-
- A. Zunger, Inverse design in search of materials with target functionalities. Nat. Rev. Chem. 2, 1–16 (2018).
-
- T. Sousa, J. Correia, V. Pereira, M. Rocha, Generative deep learning for targeted compound design. J. Chem. Inf. Model. 61, 5343–5361 (2021). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
